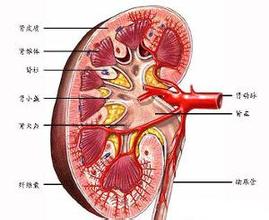
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰百科
慢性腎功能衰竭(chronicrenalfailure,CRF)是指各種腎臟疾病引起的緩慢進行性腎功能損害,最後導致尿毒癥和腎功能完全喪失,引起一系列臨床癥狀和生化、內分泌等代謝紊亂組成的臨床綜合征.從原發病起病到腎功能不全的開始,間隔時間可為數年到十餘年.慢性腎功能衰竭是腎功能不全的嚴重階段.
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰病因
腎小球腎炎(30%):
慢性腎功能衰竭的病因以各種原發性及繼發性腎小球腎炎占首位,其次為系統先天畸形(如腎發育不良,先天性多囊腎,膀胱輸尿管反流等),遺傳性疾病(如遺傳性腎炎,腎髓質囊性病,Fanconi綜合征等).
全身性系統疾病(30%):
以腎小動脈硬化,高血壓,結締組織病等多見,近年來,CRF的原發病有所變化,腎間質小管損害引起的CRF也逐漸受到人們的重視,糖尿病腎病,自身免疫性與結締組織疾病腎損害,引起的CRF也有上升趨勢,在西方國傢繼發性因素已占主要原因.
糖尿病(20%):
根據美國近年的統計,引起慢性腎功能衰竭的首要疾病為糖尿病,高血壓,腎小球疾病占第3位,而我國仍以慢性腎小球腎炎為主,繼發性因素引起的CRF依次為高血壓,糖尿病和狼瘡性腎炎,另外,乙肝相關性腎炎導致的CRF也為國內外學者所關註.
發病機制
就慢性腎臟病進展,CRF的發病機制,歷年來先後提出“尿毒癥毒素學說”,“完整腎單位學說”,“矯枉失衡學說”,“腎小球高濾過學說”,“脂質代謝紊亂學說”,“腎小管高代謝學說”等等,但沒有一種學說能完整地解釋其全部的發病過程,近十餘年來,隨著分子生物學的飛速發展及其在腎臟病領域的應用,加深瞭人們對CRF發生機制的認識,已有的學說不斷得到補充和糾正,新的學說不斷湧現,特別是逐漸認識瞭各種生長因子和血管活性物質在CRF進展中的作用,晚近,有學者又提出瞭“尿蛋白學說”,“慢性酸中毒學說”以及高蛋白飲食,腎內低氧對腎功能的影響,對於瞭解腎小球疾病如何引起腎小管,間質損害,腎小管,間質損害又如何加重腎小球疾病的認識具有重要意義.
腎小球高濾過學說:20世紀80年代初,Brenner等在5/6腎切除大鼠,應用微穿刺研究證實殘餘腎的單個腎單位腎小球濾過率(singlenephronGFR,SNGFR)增高(高濾過),血漿流量增高(高灌註)和毛細血管跨膜壓增高(高壓力)即著名的“三高學說”或“腎小球高濾過學說”.
其產生的機制主要是殘餘腎單位入球小動脈較出球小動脈擴張更加顯著所致,一般認為是因為入球小動脈擴張與擴血管物質前列腺素分泌過多,以及對血管緊張素Ⅱ(AngⅡ)不敏感有關,而出球小動脈擴張相對較少則與該動脈對AngⅡ的敏感性增加有關;另外,入球小動脈對AngⅡ的敏感性低與局部內皮細胞來源血管舒張因子(EDRF,現認為主要是NO)分泌增多有關.
當處於高壓力,高灌註,高濾過的血流動力學狀態下,腎小球可顯著擴展,進而牽拉系膜細胞,周期性機械性牽拉系膜細胞,可以使膠原Ⅳ,Ⅴ,Ⅰ,Ⅱ,纖維連接蛋白(fibronectin,FN)和層粘連蛋白(laminin)的合成增多,細胞外基質(ECM)增加,腎小球肥大在某種程度內得到緩沖並減輕瞭腎小球壓力,增加腎小球順應性,然而,大量ECM積聚,加以高血流動力學引起腎小球細胞形態和功能的異常,又會使腎小球進行性損傷,最終發展為不可逆的病理改變即腎小球硬化.
矯枉失衡學說:20世紀60年代末~70年代初,Bricker等根據對CRF的一系列臨床和實驗研究,提出瞭矯枉失衡學說(trade-offhypothesis),這一學說認為,CRF時體內某些物質的積聚,並非全部由於腎臟清除減少所致,而是機體為瞭糾正代謝失調的一種平衡適應,其結果又導致新的不平衡,如此周而復始,造成瞭進行性損害,成為CRF患者病情進展的重要原因之一.
時,甲狀旁腺激素(parathyroidhormone,PTH)升高造成的危害是最好的說明,隨著CRF降低,尿磷排泄量減少,引起高磷血癥,由於血清中鈣磷乘積的升高,一方面使無機鹽在各器官(包括腎臟)沉積,出現軟組織鈣化;另一方面,低鈣血癥又刺激瞭PTH的合成和分泌,以促進尿磷排泄,並升高血鈣,但對甲狀旁腺的持續性刺激可導致甲狀旁腺的增生及繼發性甲狀旁腺功能亢進(secondaryhyperparathyroidism,SHP),從而累及骨骼,心血管及造血系統等.
矯枉失衡學說對於進一步解釋各種慢性腎臟疾病進展的原因,加深人們對CRF時鈣磷代謝紊亂及SHP發病機制的認識具有重要意義,一直為各國學者所推崇,近30年來,這一領域的研究取得瞭重大進展,對Bricker等提出的這一學說的某些認識又有瞭新的認識.
首先,磷的瀦留並非產生SHP的始動因素,大量研究報告表明,在腎衰早期患者血清PTH升高之前,並未出現高磷血癥,血磷水平反而降低,隻有當腎衰進入晚期(GFR<20ml/min)時,患者才出現磷的瀦留,而高磷血癥不僅通過低鈣血癥,亦可以通過其它途徑或直接促進PTH的分泌,近年來發現,腎小管細胞內磷負荷升高,抑制瞭1α-羥化酶的活性,使25-(OH)2D3轉化為1,25-(OH)2D3減少,削弱瞭對PTH分泌的抑制,磷對甲狀旁腺還可能具有直接作用,因為低磷飲食可在血清中鈣和1,25-(OH)2D3濃度無變化的情況下,降低PTH及其前體PTHmRNA的水平.
其次,低鈣血癥並非引起SHP的惟一直接原因,實際上,早期腎衰患者在低鈣血癥出現之前血清PTH已經升高,補充血鈣至正常水平並不能阻止SHP的產生與發展,因此,除瞭低鈣血癥外,還有其他重要因素參與瞭SHP的形成.
腎小管高代謝學說:研究認為,在慢性腎衰進展過程中,腎小管並不是處於被動的代償適應或單純受損狀態,而是直接參與腎功能持續減低的發展過程,其中,腎小管高代謝已為動物實驗所證實,當大鼠切除5/6腎後,其殘餘腎單位氧耗量相當於正常大鼠的3倍,其機制可能是多方面的,如可能與殘餘腎單位生長因子增加,溶質濾過負荷增加,脂質過氧化作用增強,多種酶活性增加,Na-H反向轉運亢進和細胞內Na流量增多有關.
腎小管的高代謝可引起剩餘腎單位內氧自由基生成增多,自由基清除劑(如谷胱甘肽)生成減少,進一步引起脂質過氧化作用增強,進而導致細胞和組織的損傷,使腎單位進一步喪失.
此外,間質淋巴-單核細胞的浸潤並釋放某些細胞因子和生長因子,亦可導致小管-間質損傷,並刺激間質纖維母細胞,加快間質纖維化的過程.
蛋白尿學說:近年來,尿蛋白在腎小管-間質損害中逐漸引起人們的重視,臨床和實驗研究均證實尿蛋白作為一個獨立的因素直接同腎功能損害程度正相關.
臨床上,ACEI不僅能控制腎病患者高血壓,而且能降低蛋白尿,即使對血壓正常的患者亦有此作用,並且有延緩腎功能下降趨勢,進一步支持以上結論,但尿蛋白如何加重腎功能損傷的機制尚未真正闡明,可歸納為以下幾個方面:
尿蛋白對腎小球系膜細胞的毒性作用:大多數進行性腎功能衰竭動物模型都可觀察大量蛋白在系膜區積聚,可以促進系膜細胞增生,增加ECM蛋白產生,因而加重腎小球硬化,特別值得註意的是,脂蛋白在此過程中起著重要的作用,動物實驗表明,在蛋白尿狀態下,腎小球內有大量脂蛋白如ApoB,LDL,VLDL及ApoA積聚,脂蛋白可以引起下面一系列變化:
①LDL同其系膜細胞上受體結合,刺激原癌基因如c-fos和c-jun表達,導致系膜細胞增生.
②LDL可以增加ECM蛋白中糖蛋白如FN產生,誘導MCP-1和PDGF基因表達增加.
③LDL可以在巨噬細胞和系膜細胞中形成氧化型LDL,現在認為氧化型LDL比LDL更具有毒性,可以刺激巨噬細胞產生多種生長因子,細胞因子和其能刺激膠原合成和系膜細胞增殖的介質,進一步促進腎小球硬化,給予抗氧化劑如維生素E和維生素C能明顯降低氧化型LDL的毒性作用.
尿蛋白對近端腎小管細胞的直接毒性作用:正常情況下,腎小球濾過的蛋白質可以出現在腎小管液中,再通過入胞作用在近端小管重吸收入血,但大量蛋白質超過腎小管重吸收能力,可以引起腎小管的損害,過度的尿蛋白可以增加溶酶體的負荷,引起溶酶體腫脹,破裂,大量溶酶體中蛋白酶釋放入血中,引起近端腎小管損傷.
尿蛋白可以改變腎小管細胞生物活性:從胚胎來源上,近端腎小管來源於間充質細胞,與成纖維細胞和免疫系統的細胞接近,最近的研究表明尿蛋白可以調節腎小管細胞功能,改變它們的生長特性和細胞因子及基質的蛋白的表型.
細胞培養研究證實,當人類腎臟皮質上皮細胞暴露在腎小管液環境中,可以使MCP-1mRNA和蛋白表達增加,MCP-1主要為單核細胞產生,亦可由腎小管上皮細胞產生,它可以不依賴於酪氨酸激酶或蛋白激酶途徑,本身具有獨特的信號轉導通路,即借助轉錄因子-核因子卡巴B(nuclearfactorkapaB,NFκB),通常情況下,NFκB以無活性形式存在於細胞漿中,可被其抑制亞單位IκB蛋白降解產物激活,結果,NFκB二聚體轉位至細胞核內,充當轉錄因子刺激幹擾素,細胞介質及細胞黏附因子等基因轉錄,包括MCP-1.
蛋白尿介導腎小管損害亦與整合素表達有關,後者是一種介導細胞與細胞,細胞與ECM黏附的異二聚體糖蛋白,在ECM蛋白合成,降解及再分佈方面起著重要的作用,正常情況下,在人類培養的腎小管細胞上普遍表達α3β1,局限性表達αvβ5,最近的研究表明當培養的腎小管細胞中加入白蛋白時,可引起一個劑量依賴的αvβ5表達,隻是這種白蛋白需攜帶脂分子才有此作用.
一些特殊的蛋白質引起的腎損害:白蛋白在腎小管液中可引起損傷,這種脂分子主要是其中含有的脂肪酸引起.
腎小球濾過液轉鐵蛋白流過腎小管時,其酸性環境可以使轉鐵蛋白釋放出Fe2離子,Fe2離子可引起腎小管細胞釋放乳酸脫氫酶(LDH)和脂質過氧化物丙二醛,通過氧自由基損害腎小管,亦有研究證實,轉鐵蛋白可以使近端腎小管細胞MCP-1mRNA表達上調,加重腎小球損害.
補體濾出到腎小管中,含有大量膜攻擊復合物C5b-9,在進行性腎小球損害中起著一定的作用,腎小球腎炎時,尿中氨的水平同尿蛋白水平呈正相關,大量尿蛋白重吸收產生,氨產生增加,氨可以通過旁路途徑激活補體,產生C5a和C5b-9,C5b-9則可以促進腎小球上皮細胞產生細胞介質,刺激膠原合成,引起腎小球進行性硬化.
尿蛋白對腎小球代謝的影響:綜上所述,尿蛋白在進行性腎臟損傷的機制可歸納.
脂質代謝紊亂學說:進行性腎功能損害常表現有脂質代謝紊亂,如血漿三酰甘油,極低密度脂蛋白(VLDL),低密度脂蛋白(LDL),飽和脂肪酸增多,特別是富含載脂蛋白(ApoB)的脂蛋白增多,而高密度脂蛋白和不飽和脂肪酸降低,脂質代謝紊亂除引起動脈硬化而加速腎功能損害外,還可通過多種途徑促進腎小球硬化,進一步導致腎功能進行性下降.
酸中毒矯枉失衡學說:腎臟是機體調節酸堿平衡最重要的器官之一,慢性腎臟疾病由於多種途徑異常導致腎臟對酸負荷調節能力下降,然而,對整個腎臟而言,部分健存的腎單位必然會通過多種機制加速酸性物質的產生,在一定的時間內,往往會維持相對正常的酸堿平衡,但是這要付出一定的代價,甚至會促進腎臟病進展,同矯枉失衡學說一樣,有學者亦把因酸中毒代償引起的腎臟損害稱之為酸中毒矯枉失衡學說(Trade-offhypothesisinacidosis),氨產生過多及酸中毒可以通過多種機制促進腎臟病進展.
氨的促生長作用:一方面氨能夠增加AngⅡ促進二酰甘油(DAG)生成的效應,氨亦能與各種生長因子協同刺激三磷酸肌醇通路,增加PKC的活性,促進蛋白質合成,另一方面氨能夠抑制蛋白質降解.
補體機制:氨可以通過激活補體的旁路途徑引起腎小管-間質損害,例如氨能直接裂解途徑中C3分子中硫脂鍵形成酰胺化C3,後者再通過C3/C5轉化酶,進而裂解C3,激活的C3可直接與系膜表面氨基起反應引起損害;亦能產生C5a和C5b-9,C5a作為趨化因子吸引各種炎癥細胞在腎小管-間質積聚,C5b-9則作為膜攻擊復合物直接溶解細胞膜.
尿鈣排出增加:枸櫞酸分子中含有3個COOH可代謝成HCO3-而抵消一部分酸負荷,然而,酸中毒時使尿枸櫞酸水平降低,而正常枸櫞酸在尿中可與鈣結合成可溶性形式排出,這樣結果必然會促進腎結石和腎石病進展,加重腎功能障礙.
促進腎臟囊腫形成:酸中毒可引起細胞內低鉀,後者促進腎囊腫形成.
蛋白質飲食與腎功能進展:高蛋白飲食引起或加重腎功能進展的機制主要有以下幾方面:
血流動力學機制:以往認為高蛋白飲食引起跨腎小球毛細血管壓升高主要是由於前列腺素代謝異常而致擴血管前列腺素如PGE2和PGI2增加所致,現在人們在實驗中發現高蛋白飲食引起的血流動力學紊亂引起的腎臟損害如腎組織肥大是不均勻分佈的,主要集中在尿液濃縮區域如外髓質的內層(IS)和近髓質腎單位,進一步研究證實一次高蛋白飲食後2~3h,血漿抗利尿激素(ADH)水平上升約2倍,同時尿滲透壓亦明顯增加,長期高蛋白飲食則血漿ADH水平約增加2.6倍,因此,現在認為高蛋白飲食所致血流動力學損害同尿液濃縮過程相近似,一方面血漿ADH升高引起髓襻升支粗段(TAL)NaCl重吸收增加,導致TAL和IS段肥大,同時增加髓質間質溶質梯度,增加尿液濃縮能力,自由水清除率下降,另一方面,TAL段NaCl重吸收增加,流向致密斑NaCl濃度下降,抑制局部RAS系統,進而抑制管-球反饋(TGF),腎小球入球小動脈擴張,GFR增加,長期腎小球高濾過可致腎臟肥大.
非血流動力學機制:高蛋白飲食還能增加近端腎小管:Na/H逆向轉運蛋白活性和氨的產生,而進一步促進腎臟肥大.
高蛋白飲食與RAS:高蛋白飲食不僅能激活全身RAS系統,亦能激活腎局部RAS系統,眾多臨床和實驗中證實一次高蛋白飲食後血漿腎素活性,血漿AngⅡ濃度以及腎臟腎素mRNA表達明顯增加蛋白飲食後腎皮質,腎小管刷狀緣ACE活性明顯增加,AngⅡ現在被認為不僅是一個血管活性物質,促進腎小球高濾過,更是一種促生長因子,可以通過多種途徑促進腎臟病進展.
色氨酸代謝產物的作用:色氨酸代謝產物硫酸吲哚酚(indoxylsulfate)亦能引起或加重腎小球硬化,正常情況下色氨酸在腸道中大腸埃希桿菌的作用下代謝為吲哚,在大腸中吸收入血,在肝臟中轉化為硫酸吲哚酚,由腎臟排泄,腎功能不全時,硫酸吲哚酚在體內積蓄,不僅作為一種尿毒癥毒素引起一系列尿毒癥癥狀之外,還能刺激腎組織產生TGFβ,TIMP和1α(Ⅳ)型膠原,促進腎臟纖維化.
精氨酸及其代謝產物的作用:L-精氨酸在內皮型NO合成酶(Enos)作用下產生的NO可擴張血管,有一定腎臟保護作用,但產生量相對較少,L-精氨酸在組織誘生型NO合成酶(Inos)作用下產生的NO,在某些腎臟病如膜增生型腎小球腎炎中則有明顯的損害作用,最近有學者在ATS誘導的腎炎模型中發現預先用NO合成酶抑制劑L-NMMA則系膜細胞溶解被抑制90%以上,同時間質單核巨噬細胞浸潤亦明顯減少,然而,在其他腎臟病如糖尿病腎病晚期NO則起明顯的保護作用.
腎內低氧與慢性腎臟病進展:由腎小球損害引起的腎內低氧的原因主要是繼發於腎小球損害所導致的腎內血流動力學紊亂,一方面,殘存的腎小球往往處於高濾過狀態,入球小動脈和出球小動脈常代償性擴張,加上原來的系統性高血壓,結果腎小球毛細血管網向腎小管間質毛細血管發生壓力性傳遞,從而引起球後毛細血管內皮細胞損傷;另一方面,對於增生性腎小球疾病,會引起腎小球毛細血管網阻塞,也會間接地影響腎小管間質毛細血管網.
此外,由於繼發於腎小球損害引起的蛋白尿,腎小管細胞重吸收尿蛋白增加,會相應地增加腎小管間質耗氧量,從而加重腎內低氧血癥.
低氧可以誘導的各種損傷介質,血管內皮細胞生長因子(VEGF),PDGF,胎盤生長因子(PGF),TGF-β1,白介素-1,6,8(IL-1,6,8)等.
尿毒癥毒素學說:早在100餘年前人們已經認識到尿毒癥中毒癥狀可能與體內產生“尿毒癥毒素”有關,後來逐漸形成瞭所謂“尿毒癥毒素學說”,當CRF進行性加重時,體液中約有200多種物質的濃度比正常增高,由於尿毒癥癥狀復雜,涉及體內各個方面,至今不能用一種或一組“毒性”物質在體內積聚來解釋尿毒癥的所有癥狀,傳統上,尿毒癥毒素仍分為以下三類:
小分子物質:
尿素是蛋白質代謝產物,分子量是0.06kD,尿素的神經毒性與其代謝產生氰酸鹽(cyanate)有關,後者可與氨基酸N端結合,改變細胞或酶3級結構及破壞其活性,如氰酸鹽可以引起神經蛋白的氨甲酰化(carbamylation),因而幹擾高級神經中樞的整合功能.
肌酐在體內合成,尿毒癥時在體內瀦留,一般低濃度的毒性作用不大,但在達到一定濃度時,肌酐能引起細胞壽命縮短,進而溶血,肌酐還可以引起嗜睡,乏力等神經肌肉系統的功能異常.
尿酸是嘌呤的代謝產物,為水溶性化合物,尿酸主要是引起痛風,最近認為尿酸能幹擾1,25-(OH)2D3的產生和代謝,CRF患者給予別嘌醇後不僅能降低血尿酸水平,而且能提高1,25-(OH)2D3水平,尿酸與CRF患者1,25-(OH)2D3抵抗亦有一定的關系.
胍類物質隻有在達到一定濃度時才會產生毒性作用,臨床發現尿毒癥患者組織中各種胍類物質濃度上升,其中甲基胍(methylguanidine),帶正電荷,易於與細胞膜系統的磷脂等結合,因此當甲基胍聚集於組織中可引起各器官系統損害,據報道,甲基胍可引起厭食,惡心,嘔吐,腹瀉,消化性潰瘍和出血,皮膚瘙癢,貧血,抽搐和意識障礙以及糖耐量異常,還會引起肺水腫,肺淤血,肺泡出血和心肌變性,心室傳導阻滯,心功能不全.
胍基琥珀酸(guanidinosuccinicacid),其毒性作用較甲基胍低,但它可以抑制血小板第Ⅲ因子的活性並可促進溶血,可能與尿毒癥時的出血和貧血有關.
酚類包括甲酚,4-羥基苯甲酸,4-羧基苯甲酸,二羧苯甲酸和酚酸,其中,酚酸是芳香族氨基酸(苯甲氨酸,酪氨酸)經脫氨基,脫羧基和氧化作用生成,是假性神經遞質,主要引起中樞神經系統的抑制作用,另外,高濃度酚類還可引起體內酶如Na-K-ATP酶,Mg2-ATP酶,Ca2-ATP酶活性抑制.
胺類包括脂肪族胺,芳香族胺和多胺,脂肪族胺來源於肌酐和膽酸代謝產物,可引起肌陣攣,撲翼樣震顫及溶血作用,還可抑制某些酶的活性.
芳香族胺為苯丙氨酸,酪氨酸的代謝產物,主要引起腦組織抑制作用.
多胺來源於鳥氨酸和賴氨酸代謝產物,高濃度的多胺類物質可引起厭食,惡心,嘔吐,蛋白尿,並對促紅細胞生成素,Na-K-ATP酶,Mg2-ATP酶有抑制作用,據報道多胺物質還能增加微循環的通透性,因此,可能與尿毒癥肺水腫,腹水,腦水腫形成有關.
另外,腸道細菌某些酶還能引起吲哚類物質增加,可能產生一定的尿毒癥毒性作用.
中分子物質:分子量0.5~5kD,Bergstrom等應用現代生化技術測得尿毒癥患者體內存在一組分子量介於小分子物質和大分子物質之間的物質,與尿毒癥某些癥狀有關,現發現這些物質主要是一些多肽類物質,主要引起周圍神經病變,尿毒癥腦病,糖耐量異常,還對細胞生成,白細胞吞噬,淋巴細胞與纖維細胞增生有明顯的抑制作用,但近年來爭論較多.
中分子學說有助於臨床工作者合理地選擇血液凈化方案,由於人腹膜對中分子物質有較好的通透性,因此對於中分子物質很高的人群,可選擇腹膜透析.
大分子物質:分子量>5kD,這些物質主要是一些內分泌激素,如生長激素(GH),甲狀旁腺激素(PTH),促腎上腺皮質激素(ACTH),胰高血糖素,胃蛋白酶及胰島素等,其中以PTH和胰島素作用更為突出.
過高可引起腎性骨營養不良,無菌性骨壞死,轉移性鈣化,皮膚瘙癢,透析癡呆,周圍神經病變,腎小管損害,還能抑制促紅細胞生成素產生並降低其活性,同尿毒癥貧血有一定的關系,此外,PTH亦能抑制肝臟脂酶活性,下調其mRNA表達以及抑制脂蛋白脂酶的活性,從而加重尿毒癥脂質代謝異常.
高胰島素血癥可引起紅細胞膜Na-K-ATP酶,Mg2-ATP酶活性下降,抑制腎小管Na-H交換,Na-K交換,同尿毒癥水鈉瀦留有一定關系,還可引起脂肪和肝細胞胰島素受體信號傳導途徑異常,加重尿毒癥糖代謝紊亂.
此外,還有若幹種低分子量蛋白質如核糖核酸酶,β2-微球蛋白,溶菌酶,β2糖蛋白等,當這些物質在體內濃度升高,均可能有毒性作用,其中β2-微球蛋白引起全身性淀粉樣病變已為人們所熟知.
時循環和組織中的晚期糖基化產物(AGE)含量明顯增加,並參與許多尿毒癥並發癥的發生,因此亦被認為是一種新發現的“尿毒癥毒素”,AGE是Maillard反應的終產物,AGE瀦留主要引起CRF的遠期並發癥,如AGE可使CRF患者血管壁膠原增加,引起血管硬化;AGE亦能修飾LDL,而損害LDL受體介導的清除機制,參與CRF脂質代謝紊亂的發生,AGE可以修飾β2-MG(β2-MG-AGE),與CRF淀粉樣病變密切相關,近期研究還證實,AGE修飾的β2-MG能促進尿毒癥骨病的發生,β2-MG-AGE能增加單核細胞趨化性,刺激單核巨噬細胞分泌IL-1β,TNFα和IL-6等促進骨吸收的細胞因子,刺激關節滑膜細胞分泌膠原酶從而增加結締組織的降解,並能促進破骨細胞的骨吸收作用,抑制成纖維細胞膠原蛋白的合成.
各種細胞介質,生長因子與腎臟病進展:促進腎臟病進展的細胞介質,生長因子可分為以下四類:
促炎癥分子:促炎癥分子最初的作用是增加局部炎癥反應,既可以通過激活補體,亦可以通過刺激或增加局部淋巴細胞和血小板聚集,例如,許多腎小球疾病由於局部免疫復合物沉積或形成可激活補體,這些補體大部分來源於血循環,少部分可以由局部合成,激活的補體成分如C5b~9功能上可看作一種“細胞介質”,刺激腎小球細胞增生,生長因子釋放,氧自由基產生和類花生四烯酸形成,其他細胞介質如IL-1,TNF-2與IFN-2等則可通過增加淋巴細胞趨化,黏附和釋放氧自由基來上調炎癥反應損害腎小球.
血管活性物質:縮血管物質有AngⅡ,ET-1和血栓素,AngⅡ對於腎臟疾病作為縮血管物質主要是優先收縮腎小球出球小動脈,增加腎小球跨毛細血管壓而損害腎小球,促進腎小球硬化,AngⅡ亦能收縮球後毛細血管床,導致局部缺血,促進腎小管-間質損害,此外AngⅡ可以作為一種生長和基質促進因子加重腎小球損害,而且不依賴於其血流動力學效應,ET-1是另一種主要的縮血管物質,可以引起腎臟血液灌註不全,降低GFR和RBF,加重多種腎臟病進展.
擴血管物質主要是起腎臟保護作用,如前列腺素和NO,研究證實應用非皮質激素類制劑可以加重腎功能不全,而給予PGE2則可以改善腎功能,減輕局部細胞介質和基質產生,在環孢素腎病模型中同樣證實腎組織NO能明顯減輕腎小管-間質損害,然而NO亦可不依賴其血流動力學效應而損害腎小球,如NO能刺激腎小管系膜細胞釋放多種細胞介質.
生長因子/基質促進物質:生長因子/基質促進物質主要介導腎組織損傷以後的過度修復,如前所說,一旦某種腎臟損傷發生之後,盡管其進展速度有可能不同,歸根到底總是要發展為進行性腎組織纖維化,腎功能喪失,其根本原因就是由於各種損傷後會激活各種生長因子/基質促進物質,致腎組織過度修復,如PDGF,bFGF,GH和IGF-1等均能直接刺激腎小球系膜細胞增生,分泌ECM等.
其中更重要的是TGF-β介導的效應,TGF-β是一種多功能的細胞介質,廣泛存在於成纖維細胞,單核細胞,血小板,血管內皮細胞,腎小球系膜細胞及腎小管上皮細胞,主要參與ECM形成過程,具體表現在:
①TGF-β能直接刺激ECM中多種成分如FN,膠原和蛋白多糖形成,現在認為這種調節主要發生在轉錄水平.
②TGF-β可以通過基質蛋白酶的介導影響基質降解過程,研究表明TGF-β可以抑制纖溶酶原激活因子的活性,並增加纖溶酶原激活因子的抑制因子-1(PAI-1)的活性,進而可以提高基質中PAI-1水平,PAI-1可以抑制纖溶酶原激活劑因子uPA和tPA的合成,後者可以使纖溶酶原轉化為纖溶酶,纖溶酶可以降解ECM中多種成分並能激活金屬蛋白酶(MMPs)活性進而降解膠原,因此,TGF-β主要通過增加PAI-I活性而抑制基質降解.
③TGF-β亦可以調節基質細胞整合素受體表達而促進細胞與基質黏附及基質沉積.
④更重要的是,TGF-β可通過自分泌作用誘導其本身的生成,從而大大地增強瞭它的生物活性.
此外,除瞭以上一系列生長因子能促進腎組織纖維的積聚外,近年來大量的研究表明AngⅡ和ET-1不僅能作為一種血管活性因子促進腎小球損傷,亦可作為一種基質促進物質加重腎組織纖維化,即所謂的非血流動力學效應,AngⅡ既能影響ECM合成又能抑制其降解,AngⅡ促進ECM積聚主要是通過TGF-β的介導,如AngⅡ通過其靶細胞上ATl受體作用誘導各種原癌基因如c-fos和c-jun表達,c-fos和c-jun相結合成AP-1樣轉錄因子促進TGF-β基因轉錄,此外,AngⅡ亦能使無活性TGF-β轉化為活性形式,當然AngⅡ的部分作用可能是通過刺激PDGF介導,AngⅡ抑制ECM降解則部分是通過TGF-β,部分是AngⅡ本身就可能促進PAI-l的合成,此外,AngⅡ還可通過其他多種機制促進腎組織損傷後纖維化,如AngⅡ刺激近端腎小管細胞產氨增加,氨即通過激活補體C5b~9發揮作用,AngⅡ亦能促進大分子物質進入系膜間質,誘導腎組織纖維化,最後,AngⅡ能刺激殘餘腎組織單核巨噬細胞過度增加並分泌TGF-β促進腎組織損害,ET-1是另一種基質促進物質,它不僅能介導AngⅡ,PDGF等的促有絲分裂效應,本身亦能經ETA受體激活一系列細胞內信號如PLC,PLD,PKC,酪氨酸蛋白,絲氨酸/氨酸激酶,p42,p44及MAPK,MAPK/ERK激酶,增加c-fos,fra-1和c-jun等基因表達,促進ECM合成,ET-1亦能介導系膜細胞與基質相互作用,如它能誘導系膜表達TGF-β,PDGF,EGF,此外,ET-1可激活局部黏附激酶和黏蛋白(paxillin)介導細胞與基質黏附及基質沉積.
與蛋白酶:雖然上面提到多種生長因子和基質促進物質能促進ECM進行性積聚,導致腎組織纖維化,CRF時亦涉及到ECM降解不足,正常情況下腎組織細胞內蛋白和ECM處在一個合成和降解的動態平衡的狀態下,在腎小球和腎小管-間質纖維化過程中,這種平衡往往被打破,即蛋白合成增加,各種蛋白酶活性下調,降解ECM蛋白的蛋白酶主要有三類即半胱氨酸蛋白酶,基質金屬蛋白酶(MMPs)和絲氨酸蛋白酶,絲氨酸蛋白酶包括纖溶酶,白細胞彈性蛋白酶和組織蛋白酶,MMPs包括:間質蛋白酶(如MMP-1,MMP-8)和Ⅳ型膠原酶(如MMP-2,MMP-9)以及基質溶解酶(stromatolysis).
每一種蛋白酶均有其特異性作用底物,其中纖溶酶原激活因子PA/MMP-2蛋白酶系統在ECM降解過程中起著關鍵的作用,此外,這些蛋白酶亦有其特異性抑制物如TIMPs和PAl-1,眾多的體外研究表明,多種腎臟病發展過程中存在多種蛋白酶活性下降,其抑制物的水平增加,其機制部分是通過TGF-β介導,部分由AngⅡ介導.
此外,增加的ECM現在被認為是一種“細胞介質”,可結合和瀦留多種生長因子,亦可能對細胞直接作用改變它們的表型.
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰症状
慢性腎功能衰竭的癥狀
代償作用代謝性酸中毒蛋白尿多尿呼吸氣為尿臭味昏迷昏睡急性溶血性尿毒綜合征面色蒼白尿路結石
慢性腎功能衰竭影響到各個系統和器官,可引起多種多樣的臨床表現,但是,在80%的腎單位喪失以前,或當GFP下降到25ml/min以前,可以沒有任何癥狀或隻有很少的生化改變,在諸如多囊腎等慢性進行性疾病中,即使GFR低於10ml/min,也可以沒有癥狀,這是由於殘存腎單位巨大的適應作用所致.
慢性腎功能衰竭晚期主要引起如下多種臨床病變:
水,電解質,酸堿平衡紊亂
腎臟的基本功能即調節水,電解質,酸堿平衡,腎功能不全時,由於其排泄或代謝功能障礙,必然會引起不同程度的水,電解質,酸堿平衡紊亂,然而,同ARF不一樣,CRF在其漫長的病程中由於機體各種代償機制,這些代謝紊亂有時顯得並不十分明顯,事實上,在輕中度CRF時,喪失部分功能的腎臟仍然較完全地排出各種外源性攝入和體內產生的物質或廢物,當正常的腎功能喪失約70%時,一般隻會出現部分水,電解質,酸堿平衡紊亂,隻有當腎功能進一步下降,以及攝入或體內產生過多的水,電解質,酸性或堿性物質才會出現明顯的臨床表現.
水代謝:腎臟通過其濃縮和稀釋功能調節體內平衡,正常情況下,即使每天攝水量少於500ml,腎臟亦會通過其濃縮功能保持體內水平衡,腎臟濃縮功能依賴其髓質解剖和物質轉運功能的完整性,CRF特別在腎小管間質被許多纖維組織所替代時,由於亨氏襻以及遠曲小管,集合管與其相應的直血管空間結構排列紊亂或各種主動轉運功能障礙,致使整個腎臟或集合管本身對ADH敏感性下降,結果腎臟髓質溶質梯度不能維持,尿液濃縮功能下降,此外,健存的腎單位為維持正常的腎血流量和溶質轉運,分泌過量的前列腺素特別是PGE2,以拮抗ADH,亦會損害腎臟濃縮功能,使水的重吸收產生障礙,腎臟稀釋功能是通過排泄過量的自由水來實現的,正常情況下,腎臟濾過液中12%~20%以自由水形式排出,輕度CRF時,由於健存腎單位保留其溶質重吸收功能而水的重吸收功能下降,自由水排泄相對於GFR的比例得以維持,結果水的排泄不至於發生困難,隻有到GFR下降為10ml/min,總自由水排泄低於2000ml/d,加上其他夾雜因素如血容量不足使GFR下降和降低遠端腎小管溶液流量時,才會出現水瀦留,因此這種情況迫切需要限制攝入,防止水過多和水中毒.
時既可以出現水瀦留,又可出現脫水,尿液稀釋功能障礙,不加區別地過量飲水及病變晚期大量腎單位萎縮會導致水瀦留的出現;而後者特別在尿液濃縮功能嚴重下降時可出現脫水,臨床表現為多尿,夜尿,夜尿是因為日間進食以及體內的代謝產物等溶質在日間已不能完全排出,而必須在夜間予以排出,當然,當患者伴有其他急性疾病或精神障礙致飲水量下降或水需求增加,如發熱或不顯性失水以及嘔吐,腹瀉亦會引起脫水,出現血容量不足,GFR下降,腎功能進一步惡化,後者又促進更多失水,加重尿毒癥,形成惡性循環,但若補水過多過快,又會出現水瀦留.
鈉代謝:腎臟維持體液平衡不僅表現在它對水平衡的調節,而且亦在於它對鈉平衡和血容量穩定性的調節,由於鈉主要分佈在細胞外液,影響細胞外容量和細胞內外水的分佈,因而,整個過程中鈉平衡起瞭十分重要的作用,在飲食鹽負荷和心血管系統穩定的情況下,腎小球濾過液中約99%鈉由腎小管再重吸收入血,其中50%~60%發生在近端腎小管,10%~20%發生在亨氏襻,10%~20%發生在遠端腎單位,但機制有所不同,結果排出體外的鈉僅占腎小球濾過液中不到1%,隨著飲食鈉負荷不同而有所升降,正常腎臟在飲食鈉負荷10~500mmol/d范圍內均能保持鈉平衡,CRF時,腎臟調節鈉平衡敏感性降低,直接導致細胞外容量的變化,盡管一部分患者由於原發病不同可出現失鹽,但CRF時主要表現為鈉瀦留,其根本原因在於GFR下降所產生鈉濾過下降.
隨著腎單位毀損,腎小球濾過鈉減少,致體鈉暫時性增多可使細胞外液容量過多,心血管負荷因而增加,通過心輸出量增加促使濾過鈉鹽代償性增加.
隨著體鈉瀦留可使機體產生多種適應性利鈉物質,抑制腎小管上皮細胞基底膜上Na-K-ATP酶活性,抑制鈉重吸收,如地高辛樣利鈉因子,心房利鈉多肽等,其中地高辛樣利鈉因子可以阻止全身各組織細胞Na-K-ATP酶.
時醛固酮產生不足或腎小管對醛固酮反應下降亦可促進利鈉,CRF時許多血管活性物質水平增加,作用於腎臟亦具有利鈉效應,例如ANP腎臟衍生物尿利鈉激素(urodilatin)作用於腎臟髓質抑制鈉重吸收;前列腺素特別是PGE2不僅能通過增加腎小球毛細血管血流量促進鈉濾過,而且可直接抑制腎小管鈉重吸收;其他血管活性物質如激肽釋放酶亦涉及到CRF時體鈉瀦留的適應性改變.
臨床上,CRF時鈉代謝異常所引起的各種表現早期主要歸為這些適應性過程,例如,隨著細胞內鈉和液體增加,細胞易呈去極化狀態,特別會引起神經肌肉功能失調,如肌痙攣和肌無力,各種利鈉物質增多亦會引起細胞功能失調,如循環中毒毛花苷樣物質還會引起細胞鈣增多,產生高血壓,因而,隨著腎功能進展,必須嚴格控制飲食攝入量以降低這些適應性過程,但因腎臟病變對鈉攝入量過多或不足調節的敏感性下降,對飲食鈉攝入必須慎重,突然增加鈉負荷會引起容量過多,發生高血壓和充血性心功能衰竭;相反,突然減少鈉攝入,特別是腎臟已產生適應性過程時可引起鈉不足.
鉀平衡:鉀是體內第二大陽離子,99%分佈於細胞內,約占3000mmol,細胞外僅含50~70mmol,正常飲食中含鉀量約50~100mmol,吸收入體內後主要進入細胞內,體內鉀的平衡依賴過多的鉀由細胞內流入細胞外,再由各排泄器官排出,腎臟是體內排泄鉀的主要器官,但腎小球濾出的鉀幾乎100%在亨氏襻以前被重吸收,尿中出現的鉀都是從遠端腎小管分泌的,另外,正常情況下予鉀負荷時,腎臟排鉀分數可達100%以上,隨著腎功能下降,隻要各種適應功能正常,其排泄分數亦明顯增加,因而,隻有在嚴重腎功能不全或突然少尿情況下,才會出現鉀瀦留.
時腎臟的適應性改變首先是啟動腎素-血管緊張素-醛固酮系統(RAAS)作用於遠端腎小管促進排鉀,隨著鉀排泄下降,腎小管上皮細胞內在特性發生改變,如Na-K-ATP酶活性增加促進排鉀,其原因與高鉀,醛固酮有關,其他體液因素如多巴胺能直接作用於遠端腎小管促進排鉀而並不依賴於醛固酮,其機制尚未明,CRF時許多非適應性改變亦會促進排鉀,如健存腎單位滲透壓負荷增加可提高遠端腎單位濾過液流量而增加排鉀;慢性代謝性酸中毒特別是CRF時合並近端腎小管碳酸氫鹽丟失過多可導致鉀重吸收下降.
表現為部分CRF患者,即使腎功能損害尚不太嚴重,可在臨床上頑固性高鉀血癥,既所謂鉀分泌障礙,這些患者往往存在鹽皮質激素產生不足或活性下降,腎上腺對腎素刺激敏感性遲鈍,如CRF合並原發性或繼發性腎上腺功能不全以及醫源性因素應用非皮質激素類抗炎藥,ACEI和肝素在不同水平抑制RAAS,一部分患者臨床上可表現為高鉀血癥伴輕度Ⅳ型腎小管酸中毒,其他如糖尿病腎病早期血胰島素水平不足不僅能降低鉀的排出,而且改變鉀細胞內外再分佈,促進高鉀血癥,循環中鹽皮質激素水平正常或輕度升高亦可發生鉀分泌障礙,此往往發生在梗阻性腎病,間質性腎炎,狼瘡性腎炎,鐮狀細胞病,淀粉樣病,腎移植排斥和遺傳性腎小管功能不全,這些患者在腎功能不全進展時,鉀排泄分數並不因適應改變而增加,提示小管功能存在固有的缺陷,其他如保鉀利尿劑,部分抗菌藥如三甲氧芐氨嘧啶亦會引起頑固性高鉀血癥,應引起重視.
時除瞭腎臟可發生適應性改變增加排鉀之外,許多腎外適應性改變亦可促進利鉀,主要在腸道黏膜特別是結腸黏膜可與腎小管上皮細胞一樣存在Na-K-ATP酶,對醛固酮發生反應,高鉀本身亦可直接刺激Na-K-ATP酶,嚴重CRF時腸道排鉀可增加到30%~70%.
盡管隨著腎功能進展腎臟排鉀下降,但各種適應性改變足以維持體鉀平衡,除非腎功能發生突然惡化,飲食鉀攝入量劇增,高鉀血癥的危險性仍較少,實際上,若GFR在10%以上,每天腎臟排鉀量仍可達50~100mmol,這時隻要一般的飲食控制,如1g/(kg·d)中度低蛋白和低鉀飲食即可維持體鉀平衡,然而,在存在高分解代謝如發熱,感染,溶血,消化道出血,組織損害,血腫,燒傷和手術等情況,腎前性GRF下降如血容量不足或充血性心力衰竭,服用各種能降低鉀排泄的藥物如保鉀利尿劑,ACEI,β-腎上腺能阻滯藥,肝素,非皮質激素抗炎藥和抗菌藥甲氧嘧啶等時,即使腎功能損害不太嚴重,亦會引起高鉀血癥,當然,若腎功能嚴重下降低於10ml/min以下,即使不存在以上誘因,亦會引起高鉀血癥.
部分慢性腎功能衰竭(CRF)的患者亦可表現為血鉀過低,主要因為攝入不足,大量使用利尿劑等,部分合並遠端腎小管酸中毒患者血鉀亦可過低,但嚴重腎衰時若合並低鉀,雖然應該補鉀,也必須特別小心,以防發生突然性血鉀過高.
磷代謝:CRF早期PTH水平即可升高,PTH可以抑制腎小管磷重吸收,並促進骨鈣釋放和腸道重吸收而緩解低鈣血癥,但是隨著腎功能進展,該作用已不是起代償作用,例如磷的水平逐漸升高可直接抑制PTH的作用進而減輕PTH的適應性改變,磷可抑制PTH的骨鈣釋放作用,幹擾腸道重吸收並使鈣鹽在骨中再沉積,磷亦可抑制腎組織中維生素D的羥化.
臨床上,磷代謝紊亂所引起的一系列表現主要由高磷血癥和繼發性甲旁亢引起,高磷本身可誘發轉移性鈣化和組織損害,皮膚和皮下組織轉移性鈣化則表現為瘙癢,角膜鈣化則引起帶狀角膜瘤,結合膜下鈣化則表現為急性刺激癥狀和“病眼”,關節周圍鈣化則導致肌腱炎和關節炎,血管壁鈣化可引起永久性缺血,其他如在心臟,肺臟,腦部鈣化則引起心臟傳導障礙,二尖瓣狹窄,限制性和纖維性肺病以及“器質性腦病”,腎組織鈣化可引起的腎臟損害並成為腎臟病進展機制之一,其他少見的轉移性鈣化則表現為軟組織壞死,瘤鈣質沉積病等,當鈣磷乘積超過60~70時轉移性鈣化危險性明顯增加,然而,即使在CRF時血鈣水平亦可較好地得到調節,因此,主要是由磷水平決定,一般認為血磷水平超過4mmol/L(12mg/dl),則表明體內磷負荷增加,當超過4~5mmol/L(12~15mg/dl)則轉移性鈣化危險性明顯增加.
繼發性甲狀旁腺功能亢進則主要引起骨營養不良,臨床上表現為近端肌病,軟組織鈣化和骨病,骨病主要包括下列一系列表現:
①骨軟化:表現為骨礦化不全,形成多種類骨質,其發生機制為低鈣,高磷,1,25-(OH)2VD3活性下降,PTH增多,其他如酸中毒,尿毒癥毒素,鋁中毒,營養不良亦有一定的關系.
②纖維性骨炎:主要由PTH引起,破骨細胞活性增加,骨鹽溶解,表現為海綿樣病,松質骨骨小梁形成.
③纖維囊性骨炎:為繼發性甲旁亢最具特征性的病變,主要是骨膜下骨吸收,可發生在手部,長骨,鎖骨和頭顱骨,臨床上表現為骨病,關節炎或關節周圍炎,近端肌無力,兒童則表現為生長發育遲緩,生化檢查發現堿性磷酸酶增加和不同程度鈣磷代謝異常,PTH水平明顯增加,尿中可出現無活性羧基末端片段和活性氨基末端片斷,間隙大量鈣化醇或甲狀旁腺次切除術後可減輕部分癥狀和體征,此外PTH水平升高同尿毒癥智力低下,識別功能下降及貧血亦有一定的關系,據報道PTH可抑制促紅細胞生成素(EPO)的產生.
鈣代謝:CRF時主要表現為低鈣,其機制十分復雜,如磷瀦留,PTH作用,尿毒癥毒素的毒性作用,腎體積減少及1,25-(OH)2VD3產生不足或活性下降等,CRF時鈣代謝紊亂主要表現為低鈣,然而,機體仍可發生各種適應性改變,使血鈣水平暫時得以維持,例如CRF早期腎臟濾過鈣下降可起一定的適應作用,但隨著腎功能下降,可逐漸減弱.
臨床上,低鈣血癥會引起神經肌肉應激性增加,是CRF患者手足搐搦等癥狀的常見原因,然而,由於鈣在酸性溶液中溶解度較高,雖然酸中毒時總體血鈣可能偏低,但遊離鈣水平尚正常,低鈣血癥癥狀可不出現,然而,一旦酸中毒較快糾正後,該系列癥狀可再出現,應引起臨床上足夠重視.
少數CRF時亦可發生高鈣血癥,大多是某些腎臟病進展的主要因素,如骨髓瘤,原發性甲旁亢,維生素D中毒,腫瘤組織異位產生PTH,牛奶堿綜合征,肉樣瘤病等,其他如CRF患者長期臥床及鋁中毒等均可引起高鈣血癥.
鎂代謝:主要是高鎂,由腎小球濾過減少引起,但在GFR下降至30ml/min之前,各種腎內外適應性改變可暫時性維持鎂的平衡,腎內適應性改變主要是降低腎小管鎂重吸收,增加鎂的排泄分數,除瞭鎂負荷增加可直接抑制腎小管鎂重吸收外,其他如滲透性利尿,酸中毒,PTH反應性下降及降鈣素等均能抑制鎂重吸收,腎外適應性改變主要表現腸道重吸收下降,主要與1,25-(OH)2VD3活性下降及尿毒癥毒素有關,其他如血鎂增加對骨組織及細胞攝鎂增多亦有一定的緩沖作用.
少數CRF時亦可表現為缺鎂,主要見於腎小管-間質性疾病,特別是順鉑,氨基糖苷類抗生素及戊胺治療的腎毒性,近年研究還發現長期飲酒者可導致可逆性腎小管鎂丟失過多.
當GFR低於30ml/min時各種適應性改變不足以對抗體內鎂的瀦留,特別進食含鎂的飲食時,可出現高鎂血癥,但一般臨床上無明顯表現,當血清鎂濃度>1.64mmol/L(4mg/dl)時可引起嗜睡,言語障礙,食欲不振;當>2.05mmol/L(5mg/dl)時可明顯抑制神經肌肉功能,出現昏睡,血壓下降,腱反射減弱和肌無力;隨著血清鎂濃度進一步升高,可出現心動過緩,房室傳導或心室傳導阻滯,嚴重者可致心跳驟停.
此外,鎂對鈣平衡和骨代謝亦有一定的影響,高鎂可直接抑制腎小管重吸收而致尿鈣增加,但高鎂又可抑制PTH分泌及其反應性而降低血鈣,亦有學者報告鎂不足可抑制PTH分泌,因而鎂對鈣的影響尚無定論,鎂對骨的影響主要是幹擾其正常礦化過程,與CRF時骨質營養不良有關.
代謝性酸中毒:CRF早期機體酸中毒並不明顯,主要由一系列腎內外代償性改變維持體液中pH值,腎內代償性改變為:
①部分健存腎單位代償性增加H排泄:可發生在近端腎小管,髓襻升支粗端和皮質集合管,前者主要是增加管腔膜Na/H逆向轉運蛋白活性,後者則是增加排泄H的A型間介細胞的數量而調節H分泌.
②殘餘腎單位氨的產生增加.
③降低枸櫞酸的排泄:正常情況下它可自由地濾過腎小球,99%在近端腎小管重吸收,24h尿中含8~10mmol,當GFR下降到10%時,尿中枸櫞酸排泄率僅輕度下降,大約含7mmol/24h,當GFR下降至正常1/10時,尿中枸櫞酸則可成比例下降,大約到1mmol,然而,血中枸櫞酸濃度並沒有明顯升高,說明瀦留的枸櫞酸大部分可被代謝,增加體內貯存堿.
④腎小管枸櫞酸重吸收增加:腎小管中枸櫞酸是以H枸櫞酸形式重吸收,並受Na/枸櫞酸協同轉運蛋白調節,CRF時健存腎單位排H和Na/枸櫞酸協同轉運蛋白活性增加有利於枸櫞酸重吸收;重吸收的枸櫞酸鹽可以被用來合成碳酸氫.
⑤部分CRF時血中醛固酮水平增加可直接或間接通過對鉀的排泄影響遠端小管酸化功能和氨的產生.
腎外代償首先是急性酸負荷時由細胞內,外蛋白緩沖,慢性酸負荷則動員體內堿貯備,主要是骨骼系統,骨骼是機體最大的堿貯備,大約99%鈣和88%碳酸鹽貯存在骨骼中,據研究,當體內H離子瀦留超過10~15mmol,大約需動員50%的骨堿貯,一方面與通常的生化反應,另一方面與骨質溶解有關,酸中毒時成骨細胞活性降低,破骨細胞活性增加,最後腎外代償還包括酸中毒時H細胞內流動增加,對急性酸負荷有一定的作用,但以增加細胞K離子濃度為代價.
臨床上,CRF時由於以上一系列適應性改變,往往酸中毒並不嚴重,HCO3-濃度得以維持,然而這是以機體一系列代償功能增加為代價,急性酸中毒,最主要的危害是心血管系統和中樞神經系統功能障礙,可產生致死性室性心律失常,心肌收縮力降低以及對兒茶酚胺反應性降低,心律失常的發生主要與酸中毒引起的細胞外K增加有關,當然,酸中毒對心肌細胞膜Na-K泵的抑制作用亦是原因之一,雖然酸中毒時腎上腺髓質釋放的腎上腺素對心臟具有正性肌力作用,但嚴重酸中毒又可阻斷腎上腺素對心臟的作用而引起心肌收縮力減弱,一般而言,在pH在7.40~7.20時,上述兩種相反作用幾乎相等,心肌收縮力改變不大;當pH小於7.20時,則由於腎上腺素的作用被阻斷而使心肌收縮力減弱.
酸中毒時血管系統對兒茶酚胺的反應性低下主要以毛細血管前括約肌最為明顯,而小靜脈變化不大,外周血管擴張,血壓輕度下降,對中樞神經系統主要是功能抑制,嚴重者可致嗜睡,昏迷,與酸中毒引起的腦組織內γ-氨基丁酸水平增加,氧化磷酸化過程減弱,ATP供應不足有關,酸中毒時在呼吸系統主要引起呼吸貯備不足,臨床表現為呼吸加深加快,此外,酸中毒可致組織氧離曲線左移,而組織氧供下降,其原因是由於酸中毒可抑制紅細胞內2,3-DPG產生,當嚴重酸中毒如pH<7.00時,還會引起膽堿酯酶活性下降,從而引起神經肌肉應激性改變.
糖,脂肪,蛋白質和氨基酸代謝障礙
糖代謝障礙:CRF糖代謝紊亂機制是多方面,幾乎牽涉到糖代謝的每一方面,但主要包括:胰島素抵抗;肝臟葡萄糖輸出增加;胰島素分泌異常;腎臟對胰島素清除率下降.
胰島素抵抗即胰島素敏感性下降可發生在CRF早期,在GFR下降到25ml/min之前,主要發生在外周組織,特別是肌肉組織,因為肌肉幾乎代謝體內糖負荷的90%以上,葡萄糖鉗鋏試驗表明CRF時肌肉組織的葡萄糖利用率下降56%以上,其機制主要有:
①胰島素對外周組織擴血管效應下降至葡萄糖,胰島素向外周組織輸送障礙.
②胰島素受體後信號傳導障礙,致胰島素刺激的葡萄糖轉運子4(glucosetransporter4,GluT4)由細胞內向細胞表面轉位(translocation)異常.
③胰島素調節的細胞內糖代謝關鍵酶活性下降致葡萄糖有氧或無氧代謝異常和糖原合成下降,如丙酮酸脫氫酶,磷酸烯醇式丙酮酸羧激酶,糖原合成酶在CRF時活性均明顯下降.
④循環中存在許多拮抗胰島素活性的物質,如遊離脂肪酸,生長激素,胰高血糖素,ET-1及尿毒癥毒素如假尿苷等.
⑤高蛋白飲食和貧血均能導致胰島素敏感性下降,隨著給予低蛋白飲食加α-酮酸及糾正貧血等胰島素敏感性亦隨之改善.
⑥酸中毒是CRF常有的異常,可導致胰島素敏感性下降,機制尚不明.
⑦CRF時各種細胞介質增多特別腫瘤壞死因子-α(TNF-α)在許多組織中均能抑制胰島素的作用.
肝臟葡萄糖輸出量增加主要表現為CRF肝臟糖異生增加,胰島對葡萄糖刺激分泌機制異常主要表現在兩方面,一方面胰島β細胞可增加胰島素分泌以克服外周組織對胰島素的抵抗,可使糖耐量試驗正常,另一方面,胰島β細胞對葡萄糖刺激的敏感性下降,使胰島素分泌減少,原因主要是繼發性甲狀旁腺功能亢進血PTH水平增加及1,25-(OH)2VD3活性下降使胰島β細胞內鈣水平增加,抑制胰島素分泌.
隨著腎功能下降,腎臟對胰島素清除率亦隨之下降,當GFR下降到40%以前,腎小管周圍細胞可增加胰島素攝取和降解維持血胰島素水平,然而當GFR下降到15~20ml/min時,最終會導致胰島素清除下降.
另外,CRF時亦可發生自發性低血糖,糖尿病患者對胰島素需求下降,主要見於外周組織對胰島素抵抗不太明顯,而腎臟對胰島素清除已明顯下降的病例,當然,CRF時長期進食不足,嚴重營養不良時低血糖亦可出現.
蛋白質和氨基酸代謝障礙:CRF患者常表現有蛋白質,氨基酸合成下,分解代謝增加及負氮平衡,若不及時糾正,在兒童可出現生長發育遲緩,成人則表現為蛋白質營養不良,嚴重影響患者康復,傷口愈合並增加感染機會,是CRF患者發病率和死亡率增加的重要因素,除瞭厭食和長期低蛋白飲食可引起蛋白質代謝障礙外,CRF發病過程本身固有的病理生理改變也是引起或加重蛋白質代謝障礙的重要因素,主要有代謝性酸中毒,胰島素抵抗,繼發性甲狀旁腺功能亢進,皮質激素水平增加,尿毒癥毒素及IGF-1抵抗和一些細胞介質等.
代謝性酸中毒可伴隨於CRF全過程,一方面可增加支鏈氨基酸酮酸脫氫酶活性(BCKAD),促進支鏈氨基酸(BCDA)分解,另一方面可激活促進蛋白質降解各種酶系統特別是泛素-蛋白質降解小體途徑(ubiquitin-proteasomepathway,UPP),進一步促使蛋白分解增加.
各系統功能障礙
消化系統:消化系統癥狀是CRF最早和最突出的表現,常為CRF的診斷線索,早期表現為厭食,食後胃腸飽脹感,隨著腎功能進展,特別是尿毒癥期間可出現惡心,嘔吐,腹瀉,嚴重者可致水,電解質和酸-堿平衡紊亂,加重尿毒癥癥狀,形成惡性循環,口腔炎,口腔黏膜潰瘍在尿毒癥時亦不少見,患者可有口臭,帶氨味,腮腺常腫大,食管黏膜可有灶性出血,大部分患者還可出現胃或十二指腸潰瘍癥狀,經內鏡證實潰瘍病發生率可達60%以上,胃和十二指腸炎亦十分常見,癥狀常與潰瘍混淆.
此外,上消化道出血在尿毒癥人群中十分常見,可出現嘔血,黑便,嚴重者可致大出血約占尿毒癥死亡總數之5%,其原因除瞭與胃腸淺表黏膜病變,消化性潰瘍,胃和十二指腸血管發育不良有關外,CRF時血小板功能障礙,血管壁硬化及凝血機制異常亦會或多或少地引起和加重上消化道出血傾向.
心血管系統:心血管系統疾患是CRF常見並發癥,亦是其進展到尿毒癥期首位死亡原因,而且並隨著腎臟替代治療的普及和發展而有所減少,一組研究表明,臨床上30%的CRF患者可有心功能不全的表現,但超聲心動圖檢查證實幾乎85%以上患者出現心臟結構的改變,另一組研究顯示尿毒癥透析患者心血管病死亡率是一般人群的20倍,腦血管病死亡率則在10倍以上,CRF心血管並發癥包括動脈粥樣硬化,高血壓,心肌病,心包炎和心功能不全,其原因主要是由CRF本身發展過程代謝異常引起,加上腎臟替代治療的伴發癥以及引起CRY之前心血管系統基礎病變.
①動脈粥樣硬化:動脈粥樣硬化是CRF,患者心血管系統異常重要表現之一,與其冠心病和腦血管意外高發率呈正相關.
合並動脈粥樣硬化發生原因包括:
機械因素:主要有高血壓和剪切力改變,高血壓在CRF患者發生率高達80%,可增加血管壁張力,促進巨噬細胞向血管內膜遷移,並直接激活壓力依賴性離子通道,還會引起血管缺血和出血.
代謝和體液性因素:包括脂肪和糖代謝紊亂,高同型半胱氨酸血癥和吸煙等,脂肪代謝紊亂除瞭本身能促進動脈粥樣硬化以外,同時被修飾的脂蛋白如氧化,氨甲酰化和蛋白質非酶糖化的脂蛋白,尤其是氧化LDL,非酶糖化晚期產物如氧化LDL-AGE不但具有脂蛋白作用而且能與血管內皮細胞AGE受體(RAGE)結合誘導血管黏附因子-1(VCAM-1)表達,促進循環中單核細胞在血管內膜聚集,高血糖和高胰島素血癥除瞭能引起脂代謝紊亂外亦可通過蛋白質非酶糖化和自身氧化產生氧自由基引起損害,高同型半胱氨酸血癥與葉酸缺乏有關,它可促進LDL自身氧化,血管內血栓形成,還可增加血管內膜細胞周期蛋白A(cyclinA)表達,刺激血管內膜細胞增生.
其他促進動脈粥樣硬化因素:如鈣,磷代謝紊亂不僅能引起動脈粥樣斑塊鈣化亦能誘導主動脈瓣鈣化,維生素E缺乏,可促使LDL自身氧化,增加血小板和單核細胞在血管內膜黏附和聚集,以及使血管平滑肌細胞增生並抑制單核細胞產生氧自由基和IL-1β,血管內皮細胞和血小板產生的縮血管物質和擴血管物質如ET-1/NO,TXB2/PGI2之間平衡失調亦可促進動脈粥樣硬化發生.
動脈粥樣硬化的結果一方面會引起動脈結構的重塑,包括彌漫性擴張,肥大和大中小動脈僵硬,另一方面可引起心臟結構的改變和心肌供血不足,如左心室肥大和心內膜下心肌血流量下降.
②高血壓:CRF患者高血壓發生率達80%,需要腎臟替代治療的患者則幾乎均有高血壓,其中3/4患者用低鹽飲食和透析除去體內過剩的細胞外液後,即能控制高血壓,另外l/4的患者用透析去除體內過剩的鈉和水後,血壓反而升高,此外,CRF患者高血壓有其固有的特征,表現為夜間生理性血壓下降趨勢喪失,部分可為單純性收縮期高血壓.
高血壓的發病機制主要有:
鈉平衡失調,致水鈉瀦留,細胞外液總量增加,使心排出量增加,繼而外周阻力增加,是CRF高血壓的首要因素,通過控制水,鈉攝入,利尿和透析可望有好轉.
內源性洋地黃類因子增加是機體對鈉瀦留的一種代償反應,可抑制腎小管上皮細胞Na-K-ATP酶,減少腎臟鈉重吸收,然而該物質亦抑制瞭血管平滑肌細胞Na-KATP酶活性,細胞內鈉水平增加,抑制Na-Ca2交換,細胞內鈣外流減少,血管平滑肌細胞鈣水平增加,導致血管平滑肌張力增加,並提高血管平滑肌細胞對縮血管物質的敏感性.
腎素-血管緊張素-醛固酮系統(RAAS)調節紊亂,僅占腎衰患者5%~10%,使用ACEI或雙腎切除,血壓可獲控制.
腎分泌的抗高血壓物質減少如PGE2,PGl2,激肽和腎髓質降壓脂質等不僅能擴張血管,利鈉排水,還能對抗RAAS作用,長期高血壓不僅能促進動脈硬化,損害心臟,亦是CRF患者腦血管意外的重要因素.
③心肌病:亦稱尿毒癥性心肌病,是指尿毒癥毒素所致的特異性心肌功能障礙,病理上特征性變化是心肌間質纖維化,發生原因有尿毒癥毒素,脂代謝障礙和肉毒堿缺乏,局部AngⅡ作用及透析相關性淀粉樣變,近年來,尿毒癥毒素中PTH被認為是尿毒癥性心肌病的重要因素,PTH不僅能引起心肌內轉移性鈣化,而且還能抑制心肌細胞膜Ca2-ATP酶,Na-Ca2-ATP酶和Na-K-ATP酶活性,促進細胞鈣負荷增多,研究還發現PTH能引起左心室肥厚,可能與細胞鈣增加或激活PKC,誘導原癌基因如c-fos,c-jun等表達有關,給予甲狀旁腺切除,1,25-(OH)2VD3和鈣通道阻滯藥等,尿毒癥性心肌病有所緩解,臨床上尿毒癥心肌病最突出的表現為左室肥厚和左室舒張功能下降,還包括充血性心力衰竭,心律失常和缺血性心臟病.
④心包炎:心包炎發生率約15.3%,可分為尿毒癥性心包炎和透析相關性心包炎,前者主要發生於透析前或透析剛開始時,由尿毒癥本身代謝異常引起,包括尿毒癥毒素,水電解質代謝障礙,繼發性甲旁亢,感染等;後者可能與透析不充分,使體液及某些毒素特別是中分子物質和PTH等蓄積有關,其他如透析過程中細胞或病毒感染,肝素應用,血小板功能低下亦有關,病理上兩類心包炎表現相似,都為纖維素性心包炎,有滲出,出血,可發展成包裹性纖維化,亞急性或慢性縮窄性心包炎,患者常有胸痛,臥位及深呼吸時加劇,發熱在“透析相關性心包炎”中較常見,心前區可聞及粗糙的心包摩擦音或捫及摩擦感,可有不同程度的心包積液體征,重癥者可發生心包填塞,這在“透析相關心包炎”中更多見的原因則與肝素過量有關,常因急性循環障礙致死,患者還可有不同程度的房性心律失常,心電圖及X線檢查可有特征性改變,血壓突然降低或透析過程中出現低血壓,是極為重要的診斷線索,尿毒癥性心包炎對加強透析治療有良好反應,對透析反應差者要考慮感染,炎癥和免疫因素,透析相關性心包炎則需改變透析治療方案如血液透析濾過,腹膜透析等.
⑤心功能不全:在CRF發展過程會發生,是CRF患者死亡的重要原因,心功能不全常表現為心悸,氣促,端坐呼吸,頸靜脈怒張,肝大及水腫,嚴重者出現急性肺水腫,透析治療常有良效,但正性肌力藥物如洋地黃類強心藥往往反應差,且易在體內蓄積中毒,改善心臟前,後負荷藥物如多巴胺,硝普鈉和酚妥拉明(立其丁)等有時能達到緩解癥狀的作用.
呼吸系統:CRF早期常可出現肺活量減低,限制性通氣障礙和氧彌散能力下降,當伴有代謝性酸中毒時可出現氣促,甚至發生Kussmaul呼吸,進入尿毒癥期,則可出現尿毒癥肺,尿毒癥性胸膜炎及肺鈣化,並且肺部感染發生率明顯增加.
尿毒癥性肺是指尿毒癥時胸部X片上呈現以肺門為中心向兩側放射的對稱型蝴蝶狀陰影,病理上主要是以肺水腫為主,肺泡上有富含纖維蛋白的透明質膜形成,主要是由於CRF時體液過多,低蛋白血癥,充血性心功能不全和尿毒癥毒素瀦留引起,特別是一些尿毒癥毒素可明顯引起肺毛細血管通透性增加,一般多見於尿毒癥晚期,臨床上常表現為咳嗽,血痰,呼吸困難.
尿毒癥性胸膜炎發生率可達15%~20%,嚴重者可出現胸腔積液,積液可呈漏出液或血性,單側或雙側可同時發生,可為多因素綜合引起,如尿毒癥毒素可使胸膜毛細血管通透性增加,充血性心力衰竭可致胸腔積液,血小板功能障礙致胸腔內出血以及血液透析時應用肝素致凝血機制障礙等等.
肺鈣化是繼發性甲狀旁腺引起的轉移性鈣化在肺部的表現,近年日益引起人們的註意,病理上可見肺泡間隔鈣質沉著,肺組織變硬,重量增加,肺泡間隔增寬進而纖維化,鈣化亦可見於支氣管壁和小動脈壁,致肺的彌散能力降低,換氣障礙及肺活量下降,臨床上主要表現幹咳,氣短,血氣分析PaO2及動脈氧含量下降,其下降程度與肺鈣化范圍或程度呈線性相關,單純胸部X線常不能清楚地顯示轉移鈣化,但亦可呈現彌漫性浸潤,常與肺水腫,感染相混淆,若進行99mTc-Diphoaphate掃描有助於鑒別診斷.
多伴有免疫功能降低,再加上貧血,營養不良,代謝性酸中毒等使機體防禦機制障礙,致CRF患者可出現各種感染,尤其是在糖尿病,膠原病,高齡和使用激素者更易發生,特別值得重視的是,近年來CRF患者肺結核發生率比一般人群增高,常伴有肺外結核如淋巴結,肝臟,骨骼及血行播散性粟粒性肺結核,若不及時治療易招致死亡,腎衰晚期接受透析後2~3月是結核病好發時期,陳舊性肺結核復發亦常見,臨床上常缺乏典型結核癥狀,可出現對一般抗生素無反應的高熱,體重減輕,食欲不振等,外周血白細胞可增加,血沉可達100mm/h以上,CRF合並肺結核時X線胸片上常無典型結核征象,痰塗片或培養檢出率亦不高,由於免疫功能低下,結核菌素試驗常呈假陰性,因而,臨床上常難以診斷,據國內學者報道應用痰結核菌PCR檢查和測定血結核菌素純蛋白衍生物(PPD)可明顯提高診斷率.
神經系統:CRF神經系統異常可分為中樞神經系統病變和周圍神經系統病變,進入尿毒癥期發生率高達86%.
中樞神經系統早期常表現為功能抑制,如淡漠,疲乏,記憶力減退,病情加重時出現記憶力,判斷力,定向力和計算力障礙,並常出現欣快感或抑鬱癥,妄想和幻覺,可有撲翼樣震顫,最後可發展為嗜睡和昏迷,病理學改變為腦實質出血,水腫或點狀出血,神經膠質細胞變性或增生,腦電圖檢查常示有明顯異常,慢波增多.
周圍神經病變常見下肢疼痛,灼痛和痛覺過敏,運動後消失,故患者常活動腿,現稱之為下肢不安綜合征(restless-legsyndrome),發生率達45%,進一步發展則有肢體無力,步態不穩,深腱反射減弱,最後則出現運動障礙,部分病者尚有自主神經功能障礙,出現直立性低血壓,發汗障礙,神經源性膀胱和早泄,病理上常表現為神經纖維脫髓鞘變,其原因為尿毒癥血中胍基琥珀酸或PTH過多,抑制神經細胞內轉酮醇酶有關,晚近,有資料表明神經纖維內鈣含量降低,可能會降低神經纖維傳導過度.
血液系統:CRF血液系統異常可表現為貧血,出血傾向及血栓傾向.
貧血可出現在所有CRF患者,但原發病不同程度有所差異,多囊腎,高血壓,腎硬化引起的貧血相對較輕,雙腎切除,伴有腎病綜合征,明顯甲狀旁腺功能亢進者貧血相對較重.
臨床上貧血的癥狀取決於貧血的程度和速度,一般主要是過度代償引起高動力學狀態的一系列表現,如心率加快,心輸出量和心搏增加,心肌前負荷和收縮力增加,長期可致心肌增厚和血管擴張,實驗檢查常為正常紅細胞正常色素型貧血,網織紅細胞計數可稍降低,有時在周圍血象中可見少數不規則的紅細胞,治療上貧血不宜糾正過快,因為機體長期處於貧血,各種細胞內酶適應地依賴無氧代謝,迅速糾正貧血不但不會使機體立即從無氧代謝轉換為有氧代謝,反而會引起許多不利負作用.
出血傾向是CRF患者常見合並癥,一般為輕度出血,主要表現為皮下瘀斑,紫癜,鼻出血和牙齦出血,重者亦可出現出血性心包炎,腹膜後,胃腸道甚至顱內出血,外科手術或創傷後出血更為常見,CRF患者出血機制尚未十分清楚,主要有血小板功能障礙如血小板第Ⅲ因子活性下降,血小板膜糖蛋白GPⅡb/Ⅱa復合物活性受損,血小板貯存缺乏及血小板產生TXA2減少,後者可能與環氧化酶活性下降有關,此外,血管壁異常如PGl2產生不足,血管性(假)血友病因子(vWF)活性下降和凝血機制異常如抗磷脂抗體及狼瘡抗凝物濃度增加等亦可促進出血,然而,CRF患者亦有血栓形成傾向,表現為透析患者動-靜內,外瘺容易阻塞,原因與某些患者血小板功能呈亢進狀態有關,其他如抗凝血酶Ⅲ和蛋白C活性下降和纖維溶解不足亦能促進血栓形成,研究表明一些透析患者循環中組織型纖溶酶原激活物(tPA)活性下降而纖溶酶原激活物的抑制物-1(PAI-1)活性增加,已知tPA/PAI-1系統是纖維溶解過程中最重要的物質.
運動系統:尿毒癥晚期常有肌病,表現為嚴重肌無力,以近心端肌肉受累為主,可有舉臂或起立困難,企鵝樣步態等表現,電生理發現肌細胞靜息電位降低,動作電位時程縮短,與細胞內離子濃度變化有關,其原因主要為1,25-(OH)2VD3不足,PTH水平增加,鋁負荷過多和營養不良等,患者可有骨痛,自發性骨折,關節炎和關節周圍炎以及肌腱斷裂等改變,兒童常有生長發育遲緩及維生素C缺乏病表現,成人亦可發生腰椎側突或脊柱後突等骨骼畸形,腎性骨營養不良極常見,除瞭鈣磷代謝紊亂,繼發性甲旁亢是主要因素之外,還與鋁負荷過多和慢性代謝性酸中毒有關.
皮膚變化:尿毒癥患者可因貧血面色蒼白或呈黃褐色,這種膚色改變一度認為是尿色素增加之故,現已證明主要是由黑色素引起,成為尿毒癥患者特有的面容,因繼發性甲狀旁腺功能亢進可致皮膚瘙癢,潰瘍及軟組織壞死,尿毒癥性瘙癢還與高濃度尿素在皮膚形成尿素霜有關.
免疫系統:CRF患者伴有感染,嚴重感染占尿毒癥死亡率為13.1%~35.7%,說明機體免疫功能異常,防禦機制低下,其原因除瞭白細胞特別是多形核白細胞(PMN)功能障礙外,還存在淋巴細胞和單核細胞功能缺陷,表現在尿毒癥患者皮膚移植存活期延長,遲發性變態反應降低,接種多種疫苗(如乙肝,流感,肺炎鏈球菌等)產生抗體低下,結核感染率高達正常人群的6~16倍,病毒感染(如乙肝,巨細胞病毒等感染)機會亦明顯增多,而且一旦感染後機體難以清除,可呈病毒攜帶者.
是機體防禦細胞感染的最主要的物質,可通過黏附,消化,氧化爆發和釋放多種蛋白酶殺滅細菌,大多數研究時PMN趨化,吞噬和殺菌功能下降,其原因包括:
①鐵負荷過多,可明顯抑制PMN吞噬功能,當血清鐵大於650?g/L時即使轉鐵蛋白飽和度下降,亦可明顯抑制PMN殺菌和氧化爆發能力,此可隨著EPO治療而改善.
②細胞內鈣增多,繼發性甲旁亢,某些透析膜等,可抑制PMN吞噬和糖代謝能力,給予1,25-(OH)2VD3和鈣通道阻滯藥可望改善.
③營養不良.
④透析期間應用生物不相容性膜,一方面可激活補體致PMN在肺臟中積聚,產生低PMN血癥,另一方面激活的PMN可高表達黏附分子Mac-l(CDllb/CDl8)使它同肺泡上皮細胞黏附增加,但低表達S-選擇素,結果PMN同血管內皮黏附功能下降,而PMN同血管內皮黏附是其殺菌的第一步,此外,PMN持續呈活化狀態亦會使吞噬功能下降.
⑤尿毒癥毒素,最近不斷有報道尿毒癥循環中存在粒細胞抑制蛋白Ⅰ,Ⅱ(GIP-I,Ⅱ)大量免疫球蛋白輕鏈,PMN細胞顆粒抑制蛋白(DIPI),血管原素(Angiogenin),泛素(ubiquitin)及P-甲酚均能抑制PMN功能.
淋巴細胞主要介導機體免疫反應,細胞免疫由T細胞介導,體液免疫主要由B細胞介導,CRF時,循環中淋巴細胞計數常減少,但CD4和CD8T細胞以及CD4/CD8比值尚正常,T細胞功能障礙主要表現在T細胞對抗原刺激增生反應缺陷,IL-2和幹擾素產生下降,還表現在T細胞受體TCR/CD3復合物下調,T細胞功能障礙常與尿毒癥毒素如PTH,胍類衍生物特別是甲基胍,LDL,PGE2,鐵負荷增多有關,盡管尿毒癥時血漿IgG,IgM和IgA等水平尚正常,但B細胞對T細胞刺激產生的抗體反應明顯低下,主要與甲旁亢,鐵負荷過多,循環中可溶性抗原和Fc受體增多有關.
內分泌系統:除腎臟產生的內分泌激素發生障礙外,性激素也時常紊亂,性功能常有障礙,女性患者可出現閉經,不育;男性患者常有陽萎,精子生成減少或活力下降等表現,血漿睪酮,雌激素和孕激素水平常降低,催乳素和黃體生成激素常增多,甲狀腺功能可有低下致基礎代謝率下降,此外,CRF患者常有體溫調節紊亂,與中樞神經系統Na-K-ATP酶活性下降有關,患者表現為正常體溫曲線下調至35.5℃,因此臨床上CRF患者若體溫大於37.5℃以上提示存在嚴重感染,需要積極治療.
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰检查
慢性腎功能衰竭檢查項目
尿常規尿滲透壓心電圖腎血流量血常規血清轉鐵蛋白血糖血液檢查放射性核素腎掃描腎功能檢查
實驗室檢查
尿液檢查:尿常規蛋白一般為()~(),晚期腎功能損害明顯時尿蛋白反見減少,尿沉渣鏡檢有不同程度的血尿,管型尿,粗大寬闊的蠟狀管型對慢性腎衰有診斷價值,尿比重降低至1.018以下,或固定在1.010左右,尿滲透壓在450mOsm/kg以下,尿中BUN,Scr水平的測定,Ccr測定,尿液濃縮-稀釋功能測定有助診斷.
血液檢查:因CRF時均有貧血,故血常規檢查對CRF有重要提示作用,血紅蛋白降低,一般在80g/L以下,重者<50g/L,為正常形態正色素性貧血,白細胞正常或降低,感染或嚴重酸中毒時白細胞可升高,血小板正常或降低,紅細胞沉降率增快,其他檢查包括血漿總蛋白,白蛋白,球蛋白及其比值測定;血電解質(HCO3-,K,Na,Ca,Mg2,P3等)水平測定,一般總蛋白<60g/L;血鈣常低於2mmol/L,血磷>1.6mmol/L,血鉀,鈉,氯,CO2CP,陰離子間隙隨病情而變化,另外,應根據病情常規做以下檢查:三酰甘油,膽固醇,高密度脂蛋白,低密度脂蛋白,載脂蛋白A,載脂蛋白B,心肌酶譜,肌酸激酶,肌酸同工酶,膽堿酯酶,乳酸脫氫酶,血糖以及pH值測定.
腎功能檢查:血肌酐(Scr),尿素氮(BUN)上升,尿液濃縮-稀釋功能測定提示內生肌酐清除率(Ccr)下降.
肝功能及乙肝兩對半檢查.
血清免疫學檢查:包括血清IgA,IgM,IgG,補體C3,補體C4,T淋巴細胞亞群,B淋巴細胞群CD4/CD8比值等.
營養不良指標檢測:測定血清總蛋白,血清白蛋白,血清轉鐵素白和低分子量蛋白,測定值下降為蛋白質-熱量營養不良的指針,血漿白蛋白水平降低是營養不良的晚期指標,血清轉鐵蛋白水平常與鐵的狀況有關,血漿水平低下可見於營養不良,但並不是衡量慢性腎衰病人營養狀態的可靠指標,低分子量蛋白如前白蛋白,視網膜結合蛋白,核糖核酸酶被認為是內臟蛋白合成的非常敏感的指標,特別對於腎功能正常者更為敏感,低血漿前白蛋白水平改變可見於血液透析營養不良的病人,極低水平的膽固醇也被認為是營養不良的指標.
影像學檢查
腎臟B超:腎皮質厚度<1.5cm,判斷CRF優於以腎臟大小為標準,如雙腎萎縮,支持終末期診斷.
其他:常規做心電圖,X線胸片,骨片及胃鏡檢查,以及某些特殊檢查如X線造影,放射性核素腎掃描,CT和磁共振等對確定腎臟的外形,大小及有無尿路梗阻,積水,結石,囊腫和腫瘤等有幫助,慢性腎衰晚期腎體積縮小(多囊腎,腎腫瘤除外)為其特征性改變.
慢性腎功能衰竭 N18.902 慢性腎衰 腎功能不全 腎衰预防
如何對慢性腎衰患者進行早期預防,並延緩慢性腎衰的病情進展,已成為各國十分關註的問題,目前提出3級預防和隨訪措施.
一級預防:又稱早期預防,是對已有的腎臟疾患或可能引發CRF的原發病因,如慢性腎炎,腎盂腎炎,糖尿病,高血壓等,進行早期普查和及時有效的治療,以預防可能發生的慢性腎功能不全.
二級預防:即防止慢性腎衰持續進展和突然加重,對慢性腎衰的患者,積極糾正脂質代謝紊亂,進優質低蛋白飲食,控制高血壓,避免加劇因素,適寒溫,避風寒,避免外感,感染,同時註意合理飲食和休息,以有效阻止病情進展,促進病情恢復.
三級預防:是對進入終末期腎功衰的患者積極治療,以防危及生命的並發癥發生,如高鉀血癥,心衰,嚴重代謝性酸中毒等,以延長患者生存期,對我國這樣一個人口眾多的發展中國傢,應加強CRF的早期預防和延緩病程進展,重視非透析治療的發展,改進和推廣,透析與移植治療應在挽救生命時采用.
追蹤隨訪:慢性腎衰竭患者必須定期隨訪,就診的頻度應據病情決定,如有否高血壓,心力衰竭及殘餘腎功能惡化的速度加快等,所有的患者至少需每3個月就診一次,就診時必須詢問病史和體檢,同時做必要的實驗室檢查,如血常規,尿常規,血尿素氮,肌酐濃度以及電解質,血清蛋白,甲狀旁腺激素,鐵蛋白,C-反應蛋白等,根據病情積極對癥處理.





























