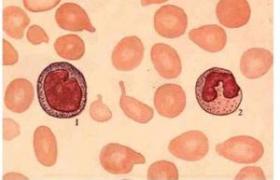
骨髓增生異常綜合征 MDS
骨髓增生異常綜合征 MDS百科
骨髓增生異常綜合征(myelodysplasticsyndrome,MDS)是一組起源於造血髓系定向幹細胞或多能幹細胞的異質性克隆性疾患,其基本病變是克隆性造血幹、祖細胞發育異常(dysplasia),導致無效造血以及惡性轉化危險性增高,主要特征是無效造血和高危演變為急性髓系白血病,臨床表現為造血細胞在質和量上出現不同程度的異常變化,MDS發病率約10/10萬~12/10萬人口,多累及中老年人,50歲以上的病例占50%~70%,男女之比為2:1.
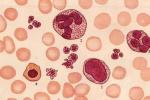
骨髓增生異常綜合征 MDS
骨髓增生異常綜合征 MDS病因
先天因素(30%)
MDS是一種源於造血幹/祖細胞水平的克隆性疾病,其發病原因與白血病類似,目前已經證明,至少2種淋巴細胞惡性增生性疾病--成人T細胞白血病及皮膚T細胞型淋巴瘤是由反轉錄病毒感染所致,亦有實驗證明,MDS發病可能與反轉錄病毒作用或(和)細胞原癌基因突變,抑癌基因缺失或表達異常等因素有關.
理化因素(10%)
患者常有明顯發病誘因,苯類芳香烴化合物,化療藥物尤其是烷化劑,放射線均可誘導細胞基因突變而導致MDS或其他腫瘤發生,此外,MDS多發生於中老年,是否年齡可降低細胞內修復基因突變功能亦可能是致病因素之一.
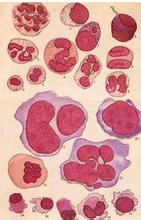
骨髓增生異常綜合征 MDS
骨髓增生異常綜合征 MDS症状
骨髓增生異常綜合征的癥狀:
鼻出血鼻衄乏力肝脾腫大關節腫痛粒細胞減少淋巴結腫大顱內出血面色蒼白內臟出血
1.癥狀MDS臨床表現無特殊性,MDS通常起病緩慢,少數起病急劇,一般從發病開始轉化為白血病,在一年之內約由50%以上,貧血患者占90%,包括面色蒼白,乏力,活動後心悸,氣短,老年人貧血常使原有的慢性心,肺疾病加重,發熱占50%,其中原因不明性發熱占10%~15%,表現為反復發生的感染及發熱,感染部位以呼吸道,肛門周圍和泌尿系為多,嚴重的粒細胞缺乏可降低患者的抵抗力,出血占20%,常見於呼吸道,消化道,也由顱內出血者,早期的出血癥狀較輕,多為皮膚粘膜出血,牙齦出血或鼻衄,女性患者可有月經過多,晚期出血趨勢加重,腦出血成為患者死亡的主要原因之一,嚴重的血小板降低可致皮膚淤斑,鼻出血,牙齦出血及內臟出血,少數患者可有關節腫痛,發熱,皮膚血管炎等癥狀,多伴有自身抗體,類似風濕病.
2.體征MDS患者體征不典型,常為貧血所致面色蒼白,血小板減少所致皮膚淤點,淤斑,肝脾腫大者約占10%左右,極少數患者可有淋巴結腫大和皮膚浸潤,多為慢性粒單核細胞白血病(CMMoL)型患者.
3.特殊類型臨床表現
(1)5q-綜合征:患者第5號染色體長臂缺失而不伴有其他染色體畸變,多發生在老年女性,臨床表現為難治性巨細胞貧血,除偶需輸血外臨床病情長期穩定,很少轉變為急性白血病,50%患者可有脾大,血小板正常或偶爾增加,骨髓中最突出的表現為有低分葉或無分葉的巨核細胞,常合並中等程度病態造血,但粒系造血正常.
第5號染色體長臂有5種重要造血生長因子基因,即IL-3,IL-4,IL-5,GM-CSF,G-CSF,同時還有GM-CSF受體基因,5q-綜合征如何影響造血生長因子對造血的調控尚不十分清楚.
(2)單體7綜合征:第7號染色體呈單體樣改變,多發生在以前接受過化療的患者,單體7很少單獨出現,常合並其他染色體畸變,孤立的單體7染色體畸變常見於兒童,可出現在FAB分型各亞型,大多數有肝脾腫大,貧血及不同程度白細胞和血小板減少,25%患者合並有單核細胞增多,中性粒細胞表面主要糖蛋白減少,粒,單核細胞趨化功能減弱,常易發生感染,單體7為一個預後不良指標,部分患者可發展為急性白血病.
(3)11q-綜合征:第11號染色體長臂丟失,大多伴有其他染色體畸變,大部分為環形鐵粒幼細胞性難治性貧血(RAS)型,有環形鐵粒幼細胞增多和鐵貯存增加,一部分為難治性貧血伴原始細胞增多(RAEB)型,臨床上RAS型患者20%有11q-,第11號染色體長臂斷裂點部位報告不一,在q14~q23之間,q14斷裂點意義不明,但已知鐵蛋白H鏈基因在q13鄰近q14處,二者之間聯系尚待研究.
(4)5q-綜合征:5號染色體長臂缺失(5q-)是MDS常見的細胞遺傳學異常之一,可見於MDS的各個亞型,5q-有兩種情況:一種是單一5q-,即5q-是惟一的核型異常;另一種是復雜5q-,即除5q-外還同時有其他染色體異常改變,由於有單一5q-的RA和RARS有其特殊臨床表現和預後,故MDS的5q-綜合征是專指這種情況.
5q-綜合征主要發生於老年女性,外周血表現為大細胞貧血,白細胞數輕度減少或正常,血小板數正常或增高,骨髓中最突出的改變是巨核細胞發育異常,分葉減少的小巨核細胞明顯增多,紅系細胞發育異常的表現有時可不明顯,可有環狀鐵粒幼細胞,患者呈慢性臨床過程,主要是頑固性貧血,出血和感染少見,一般抗貧血治療無效,但僅靠定期輸血可較長時間存活,中位存活時間可達81個月,轉白率極低.
(5)鐵粒幼細胞性貧血(sideroblasticanemia,SA):SA是一組異質性疾病,其共同特征是由於不同原因引致幼紅細胞中亞鐵血紅素(heme)生物合成障礙,致使線粒體內鐵負荷過多,形成繞細胞核排列的鐵粒,即環狀鐵粒幼細胞,SA可分為三大類:①遺傳性和先天性SA;②後天性SA;③由酒精中毒和某些藥物引起的可逆性SA,MDS的RARS屬於後天性SA,後天性SA中的一個主要亞型是原發性後天性SA(idiopathicacquiredsideroblasticanemia,IASA),Kushner等曾就文獻中和自己的IASA病例進行分析,發現:①幼紅細胞PAS染色陰性;②病程長,中位活存時間長達10年;③患者的活存曲線與正常人群相同,而不呈惡性疾患模式;④轉白率很低(7.4%),MDS的RARS是否等同於IASA,FAB分型和WHO分型中都未做特別說明,但已有作者提出RARS中有兩類情況,一類應診斷為MDS,另一類仍應診斷為SA,這兩類的鑒別點如表1所示.
(6)17p-綜合征:17號染色體短臂缺失(17p-)可發生於5%左右的MDS患者,多數由於涉及17p的非平衡易位,亦可由於-17,iso(17q)或單純17p-,17p-常合並其他染色體異常,抑癌基因p53定位於17p13,上述各種核型異常所造成的17p-,缺失區帶可不完全相同,但都包括p53基因區帶,而且70%左右的17p-綜合征患者有p53基因失活,說明另一個等位p53基因也發生瞭突變.
17p-綜合征的血液學突出表現為粒系細胞發育異常,外周血中性粒細胞有假性Pelger-Huet核異常和胞質中小空泡,這種改變也可見於骨髓中不成熟粒細胞,患者臨床上對治療反應差,預後不良.
(7)CMML:20世紀70年代初,Hurdle等和Meischer等首先報道CMML,認為它是一種慢性骨髓增殖性疾病(MPD),其特征為外周血白細胞數正常或增高,偶可有幼粒或幼紅細胞,單核細胞>0.8×109/L,骨髓有核細胞增多,可有發育異常的形態表現,以粒系增殖為主,單核細胞亦增多,Ph染色體陰性,可有脾臟腫大,後來FAB協作組因其有血細胞發育異常的形態表現,將之納入MDS作為一個亞型,但由於本病有明顯的MPD特征,這種歸類一直受到質疑,現在WHO分類方案中,將CMML改劃人新增的MDS/MPD大類中,解決瞭這一長時間以來的爭議,但確有一些MDS患者,外周血白細胞數無明顯升高(<13×109/L),而單核細胞>1×109/L,臨床上亦無肝脾腫大,骨髓中血細胞發育異常的形態表現十分明顯,完全符合MDS特征,這類患者並不具備MPD的特征,顯然不應作為CMML歸入MDS/MPD中,而仍應診斷為MDS,至於是否需在MDS單列亞型,則有待商榷.
(8)aCML:本病表現類似Ph(+)CML,外周血白細胞數明顯升高,有>10%的各階段不成熟粒細胞,但與Ph(+)CML不同的是嗜堿粒細胞無明顯增多,外周血和骨髓中血細胞發育異常的形態表現十分明顯,而且常為三系發育異常,Ph染色體和bcr-abl融合基因均陰性,臨床上對治療CML的藥物反應較差,病程進展較快,中位存活時間一般<2年,過去本病被診斷為Ph(+)CML,作為CML的一個變異型,WHO分類方案制訂指導委員會和臨床顧問委員會討論後認為,本病臨床過程並非慢性,使用aCML的病名容易引起誤解,以為它是與Ph(+)CML有關系的慢性疾病,但又未能就改換一個新的病名達成一致,最後決定沿用aCML的病名,將之歸入MDS/MPD大類之中.
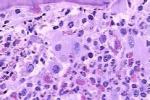
骨髓增生異常綜合征 MDS
骨髓增生異常綜合征 MDS检查
骨髓增生異常綜合征檢查項目:
染色體血清乳酸脫氫酶尿酸糖原染色葉酸心電圖骨髓增生程度血紅蛋白血清鐵血清葉酸
1.外周血:全血細胞減少是MDS患者最普遍也是最基本的表現,少數患者在病程早期可表現為貧血和白細胞或血小板減少,極少數患者可無貧血而隻有白細胞和(或)血小板減少,但隨著病程進展,絕大多數都發展為全血細胞減少,MDS患者各類細胞可有發育異常的形態改變,外周血可出現少數原始細胞,不成熟粒細胞或有核紅細胞.
2.骨髓象:有核細胞增生程度增高或正常,原始細胞百分比正常或增高,紅系細胞百分比明顯增高,巨核細胞數目正常或增多,淋巴細胞百分比減低,紅,粒,巨核系細胞至少一系有明確的上述發育異常的形態改變,常至少累及二系.
(1)紅細胞生成異常(dyserythropoiesis):外周血中大紅細胞增多,紅細胞大小不勻,可見到巨大紅細胞(直徑>2個紅細胞),異形紅細胞,點彩紅細胞,可出現有核紅細胞,骨髓中幼紅細胞巨幼樣變,幼紅細胞多核,核形不規則,核分葉,核出芽,核碎裂,核間橋,胞質小突起,Howell-Jolly小體,可出現環狀鐵粒幼細胞,成熟紅細胞形態改變同外周血.
(2)粒細胞生成異常(dysgranulopoiesis):外周血中中性粒細胞顆粒減少或缺如,胞質持續嗜堿,假性Pelger-Hǜet樣核異常,骨髓中出現異型原粒細胞(Ⅰ型,Ⅱ型),幼粒細胞核漿發育不平行,嗜天青顆粒粗大,消退延遲,中性顆粒減少或缺如,幼粒細胞巨型變,可見環形核幼粒細胞,成熟粒細胞形態改變同外周血,異型原粒細胞形態特征如下:Ⅰ型的形態特征與正常原粒細胞基本相同,但大小可有較大差異,核型可稍不規則,核仁明顯,細胞質中無顆粒,Ⅱ型的形態特征同Ⅰ型,但細胞質中有少數(<20個)嗜天青顆粒.
(3)巨核細胞生成異常(dysmegalokaryocytopoiesis):外周血中可見到巨大血小板,骨髓中出現小巨核細胞(細胞面積<800μm2),包括淋巴細胞樣小巨核細胞,小圓核(1~3個核)小巨核細胞,或有多個小核的大巨核細胞,一般的巨核細胞也常有核分葉明顯和細胞質顆粒減少的改變,淋巴細胞樣小巨核細胞形態特征如下:大小及外觀與成熟小淋巴細胞相似,核漿比大,胞質極少,核圓形或稍有凹陷,核染色質濃密,結構不清,無核仁,胞質強嗜堿,周邊有不規則的毛狀撕扯緣或泡狀突起.
3.染色體核型分析
①核型異常:已報道的MDS患者骨髓細胞核型異常,其中以-5,-7,8,5q-,7q-,11q-,12q-,20q-較為多見,
②姊妹染色單體分化(sisterchromatiddifferentiation,SCD)延遲:用BrduSCD檢測法,骨髓細胞在體外培養56h不出現SCD現象為SCD-,這是細胞周期延長的反映,經過很多作者反復證實,MDS患者有無染色體異常以及異常的類型對於診斷分型,評估預後和治療決策都具有極為重要的意義,因此,細胞遺傳學檢查必須列為MDS常規檢測項目之一,另外,根據我們的經驗,MDS患者SCD-對於預示轉化為白血病有肯定價值.
4.骨髓細胞體外培養大多數MDS患者骨髓細胞BFU-E,CFU-E,CFU-MK,CFU-GEMM集落均明顯減少或全無生長,CFU-GM的生長有以下幾種情況:
①集落產率正常;
②集落減少或全無生長;
③集落減少而集簇明顯增多;
④集落產率正常甚或增多,伴有集落內細胞分化成熟障礙,成為原始細胞集落,有作者認為前兩種生長模式提示非白血病性生長;後兩種模式提示白血病性生長,常預示轉化為白血病,以紅系受累為主的RARS其CFU-GM生長可正常.
5.生化檢查MDS患者可有血清鐵,轉鐵蛋白和鐵蛋白水平增高,血清乳酸脫氫酶活力增高,血清尿酸水平增高,血清免疫球蛋白異常,紅細胞血紅蛋白F含量增高等,這些都屬非特異性改變,對於診斷無重要價值,但對於評估患者病情有參考價值.
6.骨髓活檢:原始細胞分佈異常,在骨小梁之間有原始細胞和早幼粒細胞的聚集分佈.
7.骨髓組織化學染色:有核紅細胞糖原染色呈彌漫陽性;病態巨核細胞糖原染色呈塊狀陽性.
8.細胞遺傳學檢查:Ph1染色體陰性;可見其它染色體異常.
9.其他還有作者提出一些MDS的特殊亞型,如MDS伴有嗜酸粒細胞增多(MDS-Eo),白細胞染色質異常凝聚綜合征(abnormalchromatinclumpinginleukocytessyndrome,ACCLS)等,這類報道多是個別病例報道,是否能構成特殊亞型,尚待更多觀察.
病理檢查
①造血組織面積增大(>50%)或正常(30%~50%),
②造血細胞定位紊亂:紅系細胞和巨核細胞不分佈在中央竇周圍,而分佈在骨小梁旁區或小梁表面;粒系細胞不分佈於骨小梁表面而分佈在小梁間中心區,並有聚集成簇的現象,
③(粒系)不成熟前體細胞異常定位(abnormallocalizationofimmatureprecursors,ALIP)現象:原粒細胞和早幼粒細胞在小梁間中心區形成集叢(3~5個細胞)或集簇(>5個細胞),每張骨髓切片上都能看到至少3個集叢和(或)集簇為ALIP(),
④基質改變:血竇壁變性,破裂,間質水腫,骨改建活動增強,網狀纖維增多等.
根據病情,臨床表現,癥狀,體征選擇做B超,X線,心電圖等檢查.
骨髓增生異常綜合征 MDS预防
MDS雖然有些病例發病原因不清,但很多病例是由於生物,化學或物理等因素引起的細胞克隆性增生,因此,應采取預防措施,醫務人員應認識到濫用藥物的危害性,使用化療藥要慎重;放射治療也應嚴格把握適應證;在有關工農業生產中接觸化學品等有害物質(如苯,聚氯乙烯)時,應作好勞動保護,防止有害物質污染周圍環境,以減少MDS的發病.
(一)生活調理
非特異性的預防有增強體質的作用,飲食起居的合理安排,適當的運動如太極拳之類鍛煉,散步,可以自我調節身體的失衡,MDS與情緒密切相關,情緒樂觀,精神愉快對防病極有意義.
(二)飲食調理
飲食得宜,可以養生,延年益壽,且可防病,在疾病治療過程中或治療後,通過飲食調理可避免疾病的進一步發展或復發,而有利於身體康復.
1.註意營養合理調配飲食,對肉類,蛋類,新鮮蔬菜的攝取要全面,不要偏食.
2.忌口雞屬陽,動風,MDS虛實夾雜,邪毒內空,助火動風之品宜忌,特別是陰虛火旺,出血,痰濕交阻者尤要註意.
3.冬蟲夏草燉鴨,冬蟲夏草九,鴨75克,生薑3片,黃酒站,水200ml,適加鹽油調味,文火燉2小時,飲湯食肉,治療MDS,氣陰不足,神疲乏力,舌淡紅,脈細者.
(三)精神調理
肝氣鬱結與MDS的發病關系密切,有資料提出MDS發病前有長達半年以上的較嚴重的精神刺激,因此提倡虛懷若谷,胸襟開闊,提高修養,在疾病調治過程中亦非常關鍵.





























