線粒體腦肌病 ME
線粒體腦肌病 ME百科
線粒體肌病是指因遺傳基因的缺陷導致線粒體的結構和功能異常,導致細胞呼吸鏈及能量代謝障礙的一組多系統疾病.伴有中樞神經系統癥狀者稱線粒體腦肌病.本病為一組臨床綜合征.任何年齡均可發病,兒童和青年多見.
線粒體腦肌病 ME
線粒體腦肌病 ME病因
(一)發病原因從目前對本病的研究來看,認為本病是因遺傳基因的缺陷,患者線粒體上有著各種不同的功能異常,並由此導致臨床表現多樣性.
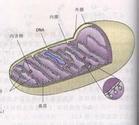
(二)發病機制已知在線粒體不同結構部位含有不同的酶系統,如外膜含有細胞色素C還原酶、脂肪酸輔酶A連接酶及單胺氧化酶;外室中含腺苷酸激酶和核苷二磷酸激酶;內膜含氧化磷酸化系統的酶類和呼吸鏈(即電子傳遞系統).氧化磷酸化要有電子傳遞.氧化磷酸化系統的酶類包括三磷腺苷合成酶、琥珀酸脫氫酶.呼吸鏈由黃素蛋白、鐵硫蛋白、輔酶Q和細胞色素所組成.此外,內膜還含有肉毒堿脂肪酸酰基轉移酶.在基質中含有檸檬酸循環酶、脂肪酸氧化酶、谷氨酸脫氫酶以及合成DNA及RNA的蛋白質結構成分.此外,人類基質中的線粒體DNA(mtDNA)也是一種遺傳物質.正是由於線粒體的結構和功能非常復雜,所以線粒體疾病在發病機制方面的“異源性”和臨床表現各異則不難理解.Jackson等(1995)分析51例線粒體肌病和腦肌病,其臨床表現同為一種綜合征或同屬線粒體肌病的臨床表現,但生化分析及分子生物學水平上的研究揭示患者在線粒體上的缺陷可以不盡相同.
肌肉的病理改變為病變肌纖維在改良Gomori三色染色切片上出現RRF,琥珀酸脫氫酶(SDH)染色呈陽性深染,SDH和細胞色素C氧化酶(COX)雙染出現藍纖維.SDH染色還可見到SDH染色強陽性血管(stronglySDH-reactivevessel,SSV),後者反映瞭血管內皮細胞或平滑肌細胞內大量線粒體積聚.COX染色可見酶活性部分或全部缺失.電鏡下可見肌膜下或肌原纖維間大量線粒體積聚,線粒體的大小和形態明顯異常,線粒體嵴內出現晶格狀包涵體,排列成停車場樣結構.此外,線粒體嵴可呈板層樣或同心圓樣排列,後者外觀似“年輪”狀.腦的基本病理改變為腦組織呈海綿狀,神經元退行性變,腦組織出現灶性壞死,星形膠質細胞增生,繼發性髓鞘脫失以及基底核鐵質沉積等.

線粒體腦肌病 ME
線粒體腦肌病 ME症状
線粒體腦肌病的癥狀傳導阻滯耳聾復視鈣化感覺障礙共濟失調呼吸困難肌張力降低肌陣攣麻痹線粒體腦肌病常見的臨床綜合征依次分述如下:
線粒體肌病(mitochondrialmyopathy):
主要表現為以四肢近端為主的肌無力伴運動耐受不能.任何年齡均可發病,兒童和青年多見.肌無力進展非常緩慢,可有緩解復發.患病幾十年後患者仍可生活自理.嬰兒線粒體肌病有嬰兒致死性和良性兩種類型.致死性嬰兒肌病多發生在出生後1周,表現為肌力、肌張力低下、呼吸困難、乳酸中毒和腎功能不全,多於1歲內死亡.良性嬰兒肌病表現為嬰兒期內肌力、肌張力低下和呼吸困難,1歲以後癥狀緩解,並逐漸恢復正常.
最常見的基因異常為mtDNA3250位點上的突變.生化缺陷主要為酶復合體Ⅰ缺乏,也可有復合體Ⅱ、Ⅲ缺乏.肌活檢可見大量RRF,血清肌酶多正常或輕度升高.可有高乳酸血癥.
線粒體腦肌病伴高乳酸血癥和卒中樣發作(mitochondrialencephalomyopathywithlacticacidosisandstrokelikeepisodes,MELAS):
是一組以卒中為主要臨床特征的線粒體病,呈母性遺傳,80%以上的患者20歲以前發病.特征性的臨床表現為反復發作的頭痛和(或)嘔吐、皮質盲(偏盲)、偏身感覺障礙.頭痛表現為偏頭痛或偏側顱面痛,反復性嘔吐可伴或不伴偏頭痛.皮質盲是本綜合征的一個非常重要的癥狀,30歲以下枕葉卒中的患者中,14%為MELAS.局限性癲癇有時是MELAS卒中發作的先兆,為本綜合征的特征之一.其他伴隨癥狀有身材矮小、智能低下、肌力減退、感音性耳聾和癲癇發作.
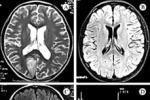
酶復合體Ⅰ缺乏是MELAS最常見(50%)的生化缺陷,此外還可有復合體Ⅲ和Ⅳ缺乏.80%的MELAS在mtDNA3243位點上有移位突變,有些患者在3271、3252、3260、3291位點上也發現瞭移位突變.MELAS主要的腦病理改變為大腦和小腦皮質、齒狀核呈海綿狀變性,大腦皮質、基底核、丘腦、小腦和腦幹多灶性壞死.大腦皮質假分層狀壞死作為缺氧性腦病的病理特征也可見於MELAS,此外腦彌漫性鈣化也很常見.由於在腦血管平滑肌、內皮細胞以及神經元細胞內均可見大量異常線粒體集聚,因此目前還不清楚卒中樣發作是由腦血管病變還是神經元功能障礙所致.肌肉活檢可見RRF和強琥珀酸脫氫酶反應性血管(stronglySDH-reactivevessel,SSV).腦CT表現為腦白質尤其是腦皮質下白質內多發性低密度灶,基底核對稱性或全腦彌漫性鈣化.
伴破碎紅纖維的肌陣攣癲癇(myoclonusepilepsywithraggedredfiber,MERRF):
為母性遺傳方式.40歲以前均可發病,10歲左右起病多見.其主要臨床特征為小腦共濟失調、肌陣攣或肌陣攣癲癇.母系親屬可呈現部分表現型,如僅有耳聾或癲癇(包括失神發作、失張力發作和強制陣攣發作).伴隨癥狀可有身材矮小、精神運動發育遲緩、神經性耳聾、視神經萎縮、眼肌麻痹、頸部脂肪瘤、周圍神經病、心臟病和糖尿病.
的生化缺陷多數為酶復合體Ⅳ缺乏,其次為酶復合體Ⅰ和Ⅳ缺乏.80%的MERRF患者在mtDNA8344位點上有移位突變.腦病理改變主要累及小腦齒狀核、紅核、殼核和Luys體.肌肉的主要病理改變為:RRF和SSV,後者反映線粒體在血管內皮和平滑肌細胞內聚集.血或腦脊液乳酸水平可升高.顱腦CT可見腦萎縮.
綜合征(KSS)及Pearson綜合征:
多在20歲以前發病,多為散發,除眼外肌癱瘓外伴視網膜色素變性和(或)心臟傳導阻滯,還可出現身材矮小、神經性耳聾和小腦性共濟失調.Pearson綜合征為一組嬰兒非神經系統紊亂癥狀,包括全血細胞下降,胰外分泌功能紊亂,肝功異常,可有腎功能衰竭,幸存者後期出現KSS表現.此二綜合征的遺傳基礎為mtDNA大量重復.
慢性進行性外眼肌麻痹(chronicprogressiveexternalophthalmoplegia,CPEO):
可為傢族性或散發性,傢族性發病的遺傳方式目前尚不能完全確定,部分為母性遺傳,也可以是常染色體顯性遺傳.任何年齡均可發病,但20歲以前發病者多見.臨床表現為眼球運動障礙、眼瞼下垂、短暫復視,多伴有易疲勞和肢體近端無力.肌活檢病理可見大量RRF和細胞色素氧化酶(COX)缺失.電鏡下可見肌膜下大量異常線粒體集聚,線粒體嵴異常和嵴內類晶體樣包涵體形成.腦脊液檢查可有乳酸增高和蛋白升高.國內學者研究證實mtDNA有雜合缺失,另經DNA測序證實mtDNA10909位點產生一個新的PvuⅡ酶切位點,且由單個堿基置換,認為是一新的點突變(陳清棠等,1996),采用蛋白A膠體金法(PGA)標記及免疫電鏡觀察,發現肌肉組織中與線粒體酶復合體Ⅰ、Ⅱ、Ⅲ及Ⅳ結合的金粒子,其程度減少,提示線粒體內呼吸鏈中的酶復合體活性降低(宋東林等,1996).
病:
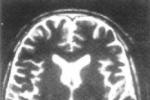
又稱亞急性壞死性腦脊髓病(subacutenecrotizingencephalomyelopathy).為傢族性或散發性線粒體腦肌病.部分為母性遺傳,部分為常染色體隱性遺傳.於出生後6個月~2歲內發病.典型癥狀為喂食困難,共濟失調,肌張力低下,精神運動性癲癇發作以及腦幹損傷所致的眼瞼下垂,眼肌麻痹,視力下降和耳聾.臨床上見到幼兒出現反復發作的共濟失調,肌張力降低,手足徐動及嘔吐癥狀應考慮此病.本病5%的基因異常與MERRF相同,為mtDNA8344和8993位點突變.腦損害分佈和病理特征與Wernicke腦病非常相似,但比Wernick腦病更廣泛,表現為丘腦、基底核、中腦、腦橋、延髓和脊髓雙側對稱性海綿狀改變伴髓鞘脫失、膠質和血管增生,周圍神經可有脫髓鞘性改變.與Wernicke腦病不同的是乳頭體很少受累.肌肉活檢除電鏡下可見線粒體增多外無其他異常.腦CT和MRI常可發現基底核和腦幹病變.血和腦脊液乳酸水平幾乎均增高.
遺傳性視神經病(Leberhereditaryopticneuropathy,LHON):
是指青春期或成年起病的急性或亞急性遺傳性視神經萎縮.1871年由Leber首次報道.男性好發,任何年齡均可發病,通常為20~30歲.臨床表現為急性或亞急性中央視野缺損.開始為單眼視物不清,數周或數月後雙眼均受累,視力損害通常較重,可致全盲.早期可有視盤水腫,萎縮期後視盤變蒼白.LHON的一個顯著特點是即使嚴重的中央視野缺損時,瞳孔對光反應仍存在.視力減退多為持續性,但相當一部分患者可有客觀的視力改善,有些甚至是戲劇性的.除瞭視覺癥狀外,還可有中樞神經的癥狀和體征、周圍神經病和心臟傳導阻滯.
的主要生化缺陷是復合體Ⅰ缺乏,基因異常為mtDNA11778位點上的移位突變,此外還有14484和3460點突變的報告.LHON的主要病理改變是視神經和節細胞層變性而不伴明顯的炎癥過程,外側膝狀體的6層均有明顯的跨神經元變性(transitionaldegeneration).肌肉活檢無RRF和SSV以及其他酶組織化學異常.
綜合征:
主要臨床表現是青少年發病的糖尿病和耳聾.此病具有發病年齡不定,程度不一,累及多器官和母系遺傳特點.遺傳基礎為mtDNA中tRNA亮氨酸(1eu)基因的3243位點發生A→G堿基置換.此病患者與MELAS綜合征的表型突變一致.
線粒體腦肌病 ME
線粒體腦肌病 ME检查
線粒體腦肌病檢查項目血乳酸肌電圖部分病人的血清CPK和(或)LDH水平升高,血乳酸和丙酮酸含量高於正常,血乳酸/丙酮酸比值升高(比值小於20為正常),均有助於診斷.
血乳酸、丙酮酸最小運動量試驗:即上樓梯運動5min後測定血乳酸、丙酮酸含量,出現含量增高及比值異常的陽性率高,對診斷更為敏感.
肌電圖:針極肌電圖多數呈肌原性損害特征.
骨骼肌活檢:
冰凍切片以改良的Gomori三色染色,在肌膜下或肌纖維內可見不規則紅色顆粒狀改變,稱破碎紅纖維(RRF),系異常線粒體堆積的一種表現.
在電鏡下可見線粒體數量增多,形態不一,有巨大線粒體,線粒體嵴排列紊亂,線粒體內可見結晶狀、板層狀包涵體,並有大量脂滴及糖原顆粒堆積.
骨骼肌呼吸鏈酶復合體活性測定可發現有異常.
外周血或骨骼肌組織mtDNA分析:可發現基因缺陷.
線粒體腦肌病 ME预防
遺傳病治療困難,療效不滿意,預防顯得更為重要.預防措施包括避免近親結婚,推行遺傳咨詢、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生.





























