
氨基酸代謝病
氨基酸代謝病百科
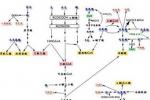
氨基酸代謝病即氨基酸病(aminoacidopathy),或稱為氨基酸尿癥,可分為兩大類:一類是酶缺陷,使氨基酸分解代謝阻滯,另一類是氨基酸吸收轉運系統缺陷.在Rosenberg和Scriver列舉的48種遺傳性氨基酸病中,至少有一半有明顯的神經系統異常,其他20種氨基酸病導致氨基酸的腎臟轉運缺陷,後者可導致繼發性神經系統損害.
氨基酸代謝病
氨基酸代謝病病因
(一)發病原因
除個別情況,氨基酸代謝病均為常染色體隱性遺傳致病,近親結婚的後代較為常見.
(二)發病機制
引起氨基酸代謝病的主要原因有兩種,即某些酶的缺乏和氨基酸的吸收障礙,前者為已知某種酶或尚不能肯定的某種酶活性缺乏或降低,如苯丙氨酸羥化酶的缺乏引起苯丙酮尿癥;分支氨基酸α-酮酸脫羧酶的缺乏或降低引起楓糖漿尿病(maplesyrupurinedisease);異戊酰輔酶A脫氫酶缺乏引起的異戊酸血癥;胱硫醚合成酶缺乏引起的同型胱氨酸尿癥;精氨酸酶缺乏引起的精氨酸血癥;賴氨酸酮戊二酸還原酶缺乏引起的高賴氨酸血癥等等,後者系由氨基酸的轉逆,吸收障礙所引起,常為腸道或其他組織對某種氨基酸的吸收障礙,如肝-腦-腎(Lowe)綜合征,Hartnup病等.
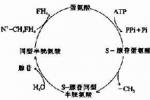
1.遺傳性酪氨酸血癥(hereditarytyrosinemia)為常染色體隱性遺傳,由15號染色體上編碼延胡索酰乙酰乙酸水解酶(fumarylacetoacetatehydrolase)基因缺陷,導致酪氨酸及代謝產物蓄積.
2.Hartnup病是中性氨基酸(如單氨酸,單羧基氨基酸等)轉運蛋白缺陷所致,該蛋白的致病基因位於2號染色體,女性攜帶致病基因傳給後代,由於色氨酸(tryptophan)通過腎小管轉運障礙,導致尿和糞便中這些氨基酸排出增多,尿中有大量尿藍母(indican)排出,主要是硫酸吲哚酚(indoxylsulfate),尤其進食含大量L-色氨酸的食物後,尿中也含大量異常的非羥化吲哚代謝產物,因大量色氨酸經尿中排出而丟失,使作為合成原料的尼克酸合成減少,導致糙皮病樣皮膚改變,本病的病理基礎尚未確定.
氨基酸代謝病
氨基酸代謝病症状
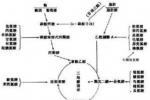
目前氨基酸代謝障礙所引起的遺傳性疾病已超過100多種,隨生物化學檢測技術的不斷進步,新發現的仍將不斷增加,氨基酸代謝病常可導致神經系統功能障礙,當神經系統受累時,通常隻出現輕度精神運動發育遲滯,直到發病2~3年後才有明顯癥狀.
像其他遺傳性代謝性疾病一樣,氨基酸病不影響胎兒的子宮內生長,發育或分娩,早期可無體征,主要臨床特征為出生時外表和活動正常,半年或1歲以後逐步出現智能減退,適當氨基酸補充,飲食控制,維生素等綜合治療後,不少病例神經癥狀可以改善.
1.遺傳性酪氨酸血癥(hereditarytyrosinemia)或稱Richner-Hanhart病,是罕見的皮膚氨基酸病(dermaticaminoacidopathy),臨床表現如下:
(1)約一半以上患兒有輕至中度智力衰退,可有自殘行為和肢體運動不協調表現,語言缺陷較突出,1歲或快滿1歲時由於角膜糜爛(cornealerosion)常引起流淚,畏光和眼睛發紅,出現新生血管形成及角膜渾濁,手掌和足底角化伴多汗和疼痛常見,是結晶酪氨酸沉積導致的炎癥反應結節,也是角膜病變的原因.
(2)可有肝,脾大或肝硬化,腹水等肝功能衰竭表現,常於患病1年或數年後死亡,新生兒期酪氨酸血癥可致肝功能衰竭和夭折,血酪氨酸及尿酪氨酸含量增高具有診斷意義,血蛋氨酸(甲硫氨酸)等氨基酸也可增高.
2.Hartnup病是較常見的色氨酸轉運障礙氨基酸病,是以第1個發病傢族的名字命名的,發病率為1/2.4萬活嬰,臨床表現如下:
(1)患兒出生時正常,嬰兒晚期或兒童早期出現癥狀,特征性臨床表現是間斷性出現紅色鱗屑狀皮疹,遍及面部,頸部,手和足等,頗似糙皮病的病損.
(2)可有生長發育遲滯,發作性人格障礙如情感多變,不能控制脾氣,精神錯亂-幻覺性精神病,發作性小腦性共濟失調(步態不穩,意向性震顫及構音障礙等),偶可出現肌痙攣,眩暈,眼震,復視和上瞼下垂等體征.
(3)日曬,情緒應激反應和服用磺胺類藥物等可激發癥狀發作,發作持續約2周,其後為一段時限不等的相對正常期,隨著患兒發育成熟,發作頻率逐漸減少,有些患兒可遺留輕度持續性智力衰退.
氨基酸代謝病
氨基酸代謝病检查
血尿便常規,肝功能,血尿氨基酸含量測定具有診斷意義.
1.遺傳性酪氨酸血癥患兒血酪氨酸含量增高,尿酪氨酸亦增高,血蛋氨酸(甲硫氨酸)等氨基酸也可增高.
2.Hartnup病患兒尿中性氨基酸增多,尿脯氨酸,羥脯氨酸及精氨酸含量正常.
3.腦電圖檢查.
4.基因檢測和產前診斷.
5.X線,CT和MRI檢查.
氨基酸代謝病预防
遺傳病治療困難,療效不滿意,預防顯得更為重要,預防措施包括避免近親結婚,推行遺傳咨詢,攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生.





























