原發性視網膜色素變性
原發性視網膜色素變性百科
原發性視網膜色素變性是一種比較常見的毯層-視網膜變性,是一組以進行性感光細胞及色素上皮功能喪失為共同表現的遺傳性視網膜變性疾病,是一種比較常見的毯層-視網膜變性,也是世界范圍內常見的致盲性眼病,該病常起於兒童或少年早期,至青春期癥狀加重,視野逐漸收縮,至中年或老年,黃斑受累致中心視力減退,甚或嚴重障礙而失明.亦有少數患者發病甚晚,但絕大多數在30歲以前發病.

原發性視網膜色素變性
原發性視網膜色素變性病因
本病為遺傳性疾病,其遺傳方式有常染色體隱性,顯性與性連鎖性遺傳3種,以隱性遺傳最多;顯性遺傳次之;性連鎖遺傳最少,但傢族史陰性的散發性病例,亦占有相當數量,通過連鎖分析,在人類多條染色體上發現瞭50多個致病基因位點,近數年來,采用定位克隆和“定位候選基因"的方法,其中18個已被確定,目前認為常染色體顯性遺傳型至少有兩個基因座位,位於第一號染色體短臂與第三號染色體長臂,性連鎖遺傳基因位於X染色體短壁一區一帶及二區一帶.

原發性視網膜色素變性
原發性視網膜色素變性症状
癥狀與功能改變
1、視網膜色素變性的臨床表現
(1)癥狀:典型的視網膜色素變性患者均有夜盲,常以此為就診的主要癥狀,常始於兒童或青少年時期,且多發生在眼底有可見改變之前.開始時輕,隨年齡增生逐漸加重.極少數患者早期亦可無夜盲主訴.表現為黃昏時戶外活動困難或室內暗光下活動受限.這種進行性夜盲的發生年齡和程度不一.早期或輕者可僅有暗適應功能減退,暗適應慢、時間延長的癥狀.早期錐細胞功能尚正常,桿細胞功能下降,使桿細胞曲線終未閾值升高,造成光色間差縮小.晚期桿細胞功能喪失,錐細胞閾值亦升高,形成高位的單相曲線.夜盲還與患眼的進行性視野損害范圍和殘留視野的大小有關.絕大多數患眼中心視力正常,直到患病晚期視野縮窄至管窺狀態患者行動困難時仍維持良好的中心視覺.多數患者童年時色覺正常,其後漸顯異常.典型改變為藍色盲,紅綠色覺障礙較少.此外,患者的夜盲癥狀及眼部病變的發生、發展的早遲與此病患者的遺傳方式有一定關系,一般在常染色體顯性遺傳(autosomaldominantinheritance,AD)型的患者,其夜盲發病年齡較遲可在成人時期,常染色體隱性(autosomalrecessiveinheritance,AR)和X連鎖隱性遺傳(X-linkedrecessiveinheritance,XL)型則發病較早,病情較重.視野與中心視力:早期有環形暗點,位置與赤道部病變相符.其後環形暗點向中心和周邊慢慢擴大而成管狀視野.中心視力早期正常或接近正常,隨病程發展而逐漸減退,終於完全失明.視覺電生理:ERG無反應,尤其b波消失是本病的典型改變,其改變常早於眼底出現改變.EOGLP/DT明顯降低或熄滅,即使在早期,當視野、暗適應、甚至ERG等改變尚不明顯時,已可查出.故EOG對本病診斷比ERG更為靈敏.
(2)眼部表現:
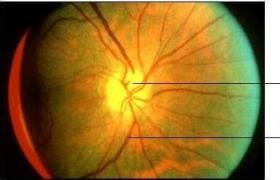
①眼底:視網膜色素變性特征性的眼底改變是視網膜色素上皮脫色素,視網膜色素上皮萎縮和色素遷移,表現為視網膜內色素沉著以及視網膜小動脈縮窄.病變的早期色素上皮損害表現為視網膜內細小的塵狀色素沉著,視網膜並因脫色素而呈蟲蝕狀或椒鹽狀外觀.隨病情進展,眼底病變從赤道部向周邊部和後極部發展,赤道及周邊視網膜出現各種形態的色素沉著,常以在血管旁聚集更為明顯.視網膜色素變性充分進展的時期,都有視網膜內骨細胞樣色素沉著和視網膜動脈血管狹窄的表現.由於視網膜色素上皮不斷脫色素和萎縮的改變,以及同時發生的脈絡膜毛細血管的逐漸萎縮,表現為脈絡膜較大血管的暴露,甚至呈現隻見脈絡膜外層大血管的脈絡膜重度萎縮的外觀.視網膜血管呈均勻一致性的白線狀血管狹窄,到晚期極細,但血管無白鞘包繞.動脈縮窄的程度較靜脈更為顯著.患眼眼底的視盤在早期正常,充分進展期則呈現典型的蠟黃色外觀.據組織病理學和視覺電生理學觀察研究,這種蠟黃的色調是神經膠質增生形成的視盤表面膜引起,並非視神經萎縮的緣故.這些研究還發現視網膜色素變性眼的視網膜神經節細胞及神經纖維層在此病過程中相對完整而很少受累.超微結構研究也證實患者的視網膜表面膜來源於視神經的星狀膠質細胞,從視盤表面向視網膜各象限伸展.
各型視網膜色素變性的早期,黃斑區外觀正常或僅見中心凹反光消失,隨後,可出現色素紊亂,中心凹旁黃斑區視網膜色素上皮脫色素.在進展期的視網膜色素變性約60%患者有萎縮性黃斑病變,約20%患者出現囊樣黃斑變性或不全性黃斑裂孔,伴放射狀內層視網膜牽引及不同程度的視網膜表面膜.大約23%患者可發生黃斑囊樣水腫.黃斑部視網膜表面膜的存在可表現為假性黃斑裂孔,在眼底檢查中應當註意.此外,大約有2%的患者可有雙側或單側視盤疣,為片層無細胞結構的鈣化物,常由神經纖維或膠質細胞圍繞,易被誤認為錯構瘤甚至視盤水腫,應註意鑒別.
②晶狀體:約50%的RP病人有後囊下白內障,表現為晶狀體後囊下後極部皮質的多孔狀或面包屑樣混濁,伴黃色結晶狀改變.最後可發展為整個晶狀體混濁,外觀似並發性白內障.故臨床上對雙眼並發白內障的患者應註意有無RP.這種病變在XL型RP中最多見.白內障形成的機制尚不清楚,有人認為與玻璃體內假炎性色素細胞有關.RP變性產物可能激活吞噬細胞,釋放活化氧分子,幹擾晶狀體代謝,或變性過程中膜脂質過度氧化產生攜帶毒性醛和脂基因的產物而直接損傷晶狀體.RP患者的白內障晶狀體超微結構的研究,僅發現可引起滲透性改變的晶狀體局部上皮變性,尚無其他特殊變化.另外RP患者也可偶發晶狀體脫位.
③玻璃體:絕大部分RP患者玻璃體可出現浮遊細胞、濃縮和後脫離.Prueff等曾將RP的玻璃體病變分為4期,即細小塵狀顆粒遍佈整個玻璃體;玻璃體後脫離;玻璃體濃縮,可伴雪球狀不透明漂浮物及玻璃體塌陷,體積顯著縮小.無論變性在任何階段,都有細小顆粒均勻分佈於玻璃體內,經玻璃體的透射電鏡檢查,發現它們為遊離的黑色素顆粒、視網膜的色素上皮、星狀細胞、類巨噬細胞及色素膜黑色素細胞.
④其他眼部表現:RP常伴有近視及散光,發生率可高達75%,尤以XL型RP中多見.據估計RP患者中青光眼發生率比正常群體中的發生率高,且主要是原發性閉角型青光眼.與RP患者易發生窄房角可能有關.另外,RP可偶爾伴有滲出性視網膜血管病變.
2、視網膜色素變性的視功能評價
(1)視力:RP早期視力相對正常或僅輕度受損.隨病情進展視力逐漸下降,絕大部分病人最後盲目.有的病人在病程後期仍保留有限的中心視力.一般認為患者一定年齡時的視力與遺傳類型有關,而中心視力下降主要是由於各種類型的黃斑病變所致.晶狀體發生的後囊下白內障也是一些RP病人視力下降的原因.
(2)色覺:RP患者最常見的色覺異常是藍色盲,而紅綠色盲較少.Fishman等對遺傳型RP患者檢查發現有萎縮性黃斑病變或視力低於20/30的患者都有色覺異常.且用Farnsworth-Munsell100色彩檢測RP患者的異常色覺比Nagel色覺鏡檢查更敏感,可發現視力尚正常的患者即有早期錐體功能受累,這種色彩分辨檢查有助於評價RP黃斑功能,並可幫助RP與錐體營養不良或錐體桿體營養不良的臨床鑒別.
二、特殊臨床類型
⑴單眼性原發性視網膜色素變性:非常少見.診斷為本型者,必須是一眼具有原發性視網膜色素變性的典型改變,而另眼完全正常(包括電生理檢查),經五年以上隨訪仍未發病,才能確定.此型患者多在中年發病,一般無傢族史.
⑵象限性原發性視網膜色素變性:亦甚少見.特點為病變僅累及雙眼同一象限,與正常區域分界清楚.有相應的視野改變,視力較好,ERG為低波.熒光造影顯示病變區比檢眼鏡下所見范圍大.本型常為散發性,但也常染色體顯性、隱性與性連鎖隱性遺傳的報告.
⑶中心性或旁中心性原發性視網膜色素變性:亦稱逆性進行性視網膜色素變性.初起即有視力減退與色覺障礙.眼底檢查可見黃斑部萎縮病變,有骨細胞樣色素堆積,ERG呈低波或不能記錄.早期以錐細胞損害為主,後期才有桿細胞損害.晚期累及周邊部視網膜,並出現血管改變.最後以失明告比利時.本型通常為隱性遺傳,偶亦有顯性遺傳.
⑷無色素性視網膜色素變性:是一種有典型視網膜色素變性的各種癥狀和視功能的檢查所見.檢眼鏡下亦有整個眼底灰暗、視網膜血管變細、晚期視盤蠟黃色萎縮等改變,無色素沉著,或僅在周邊眼底出現少數幾個骨細胞樣色素斑,故稱為無色素性視網膜色素變性.有人認為本型是色素變性的早期表現,病情發展後仍會出現典型的色素.因此不能構成一單獨臨床類型.但亦確有始終無色素改變者.本型遺傳方式與典型的色素變性相同,有顯性、隱性、性連鎖隱性遺傳三型.
目前普遍采用的RP臨床診斷標準為:①雙眼受累.②周邊視覺喪失.③桿體功能障礙:表現為暗適應桿體終閾值升高和(或)ERG桿體反應振幅降低,峰時延長或反應不能記錄.④進行性感光細胞功能喪失.而列入傳統診斷標準的典型RP眼底改變,因其隻能表示長時間的視網膜變性,在各種類型RP進展期均可見到,故不再列為RP診斷標準,尤其是對早期RP的診斷.但RP的典型眼底表現,有助於將RP與其他具有相似臨床表現但視網膜外觀不同的眼底病,如無脈絡膜癥、回旋狀脈絡膜視網膜萎縮等進行臨床鑒別.值得註意的是各種RP的最顯著特征之一為雙眼視網膜對稱性受累,在眼底表現和視功能異常方面雙眼有高度一致性.所以,臨床上對雙眼表現明顯不對稱者應考慮由其他病因引起的色素性視網膜病變.

原發性視網膜色素變性
原發性視網膜色素變性检查
基因遺傳學及免疫學檢查.
1.動態和靜態視野
Goldmann球形視野計已常規用於RP的動態視野檢查,用Ⅰ~4e,Ⅲ~4e及Ⅴ~4e視標進行檢查的結果,重復性好,相對可靠,RP早期視野表現為上方周邊視野缺損,固視點外20°~25°出現環狀暗點區,暗點區由1組孤立的暗點組成,隨病情進展暗點擴大,融合成環狀,環形暗區外緣很快向周邊擴展,而環內緣暗區則相對緩慢地呈向心性擴展侵犯,通常上方及鼻側視野首先喪失,在周邊視野完全消失後一段時間,中央視野仍可保留一小區域的黃斑視島,對AD型RP病人視野喪失速度進行研究發現,在存留10°或稍大的中央視野的患者中,小於20歲者占93%,20~40歲者占89%,而40歲以上者占60%(用Ⅳ~4e視標),Berson等對92個RP病人的視野變化進行3年的隨訪研究,發現1年後有21%的患者視野惡化,而16%的患者穩定或稍有改善,3年後則33%有惡化,14%穩定或有改善,平均每年喪失殘留視野的4.6%,隨訪過程中觀察到的視野暫時的改善可能是RP自然病程中桿體功能波動的反應,但也可能是視野這種心理物理檢查過程中的誤差,有的RP患者病情進展快,視野亦可迅速惡化,動態視野檢查對於確定視野缺損位置和范圍是簡便,有效的方法,而靜態視野對於確定視野損害的深度及特定視網膜區域的光敏感性則比動態視野更準確,正常暗適應眼的錐體對紅光比對藍光敏感,而桿體對藍光比對紅光敏感,故可用紅光及藍光光斑沿垂直或水平子午線檢查RP患者暗適應眼特定視網膜區域的閾值(即光譜敏感性),以評價患者視網膜桿體和錐體的功能,RP中常用的雙色暗適應靜態視野檢查法,一般是暗適應後用波長分別為500nm(藍綠光)及650nm(紅光)的色光斑檢測視網膜75個點,每個測試點的桿體和錐體敏感性通過與該點的正常平均值比較而得知,從2種色光刺激的敏感性異常可以確定每個檢測點的視覺由桿體和(或)錐體所介導,Massof和Finkelstein用這種雙色暗適應靜態視野檢測法分析RP患者視網膜桿體和錐體功能,發現有2種桿,錐體敏感性喪失的類型,並可據此對RP進行分型,Ernst等隨後發現瞭自動雙色靜態視野計,采用發光二極管提供紅色光及藍綠光刺激,並用此視野計對44個AD型RP傢系104個RP患者進行瞭雙色暗適應自動靜態視野檢查,進一步證實瞭AD型RP存在2種具有不同視功能損害特征的亞型.
2.暗適應閾值
檢測暗適應過程中對刺激光斑的視覺敏感性,臨床上通常用Goldmann-Weekers暗適應計繪制暗適應曲線,檢測暗適應終閾值,是評價視網膜桿體功能的敏感指標之一,RP常表現有典型的暗適應終閾值升高,但該型暗適應計僅測一固定視網膜區(常為中心凹上方11°)的局部光敏感性,近年已開始用雙色光(500nm和650nm)進行暗適應檢查,與靜態視野檢查很相似,可檢測視網膜任何部位的視覺敏感性及桿體,錐體閾值,對於評價RP患者視網膜功能是區域性受損還是彌漫性受損,以及估計視功能預後有臨床價值.
3.全視野ERG
自1945年Karpe發現RP特征性ERG表現後,隨著ERG檢測條件的改良,特別是計算機信號平均技術的應用,可檢測微伏以下的ERG反應,促進瞭RP的臨床診斷和視功能評價,采用整個視網膜均勻光刺激記錄所得的全視野ERG,通過改變刺激光波長,頻率及視網膜明,暗適應狀態,可分離出ERG-桿體和錐體反應在背景光適應下用高頻率強閃爍光刺激的錐體反應,因錐體細胞能對頻率多達70周/s的閃爍光反應,而桿體細胞僅能對至多8周/s的閃爍光反應,在暗適應下用弱藍光(<470nm)刺激則可分離出ERG桿體反應,而暗適應下用紅光刺激(>600nm)可產生分別代表桿體,錐體反應的雙峰b波,全視野ERG檢查可以評價RP中光感受器受累的類型及程度,由於桿體反應在各型RP中最早選擇性受累,全視野ERG檢查有助於RP的早期診斷,它在出現癥狀前或出現眼底可見的改變前即可表現異常;而且還可幫助識別RP傢系中的患者和正常傢系成員,RP傢系中正常與異常ERG的百分比與其遺傳型的孟德爾比例相一致,RP傢系中年齡≥6歲成員的ERG表現正常者,以後發現RP的可能性極小,ERG還可在臨床上協助鑒別早期RP與非進展性視網膜變性,前者閃爍光ERG錐體反應振幅降低,峰時延長,而後者僅表現為振幅降低,另外,全視野ERG能客觀監測RP的自然病程,為估計預後提供信息,RP患者的ERG反應常隨病情進展而變小,大多數病人ERG振幅低於0.05µV時即可成盲目,扇形RP患者ERG表現出振幅低但峰時正常者,預後好,用計算機平均的窄帶通濾過技術進行的全視野ERG檢查,還有助於客觀評價旨在穩定或延緩RP變性過程的治療嘗試,ERG在幫助識別XL型RP基因攜帶者方面也具有臨床價值.
值得一提的是ERG是代表整個視網膜外層及中層的總和電反應,通常並不與代表黃斑功能的視力成正相關,黃斑對ERG錐體反應的貢獻至多占15%左右,因此錐體反應異常並不就完全代表黃斑中心凹的損害.
4.視網膜色素變性的眼底熒光血管造影
眼底熒光血管造影可顯示RP早期用檢眼鏡查不易發現的微小RPE改變,有助於早期診斷,大多數RP病人的造影圖像顯示因RPE脫色素或RPE喪失而出現的透見脈絡膜熒光,或色素增生,沉著而引起的熒光遮蔽,15%~20%的病人有黃斑旁廣泛的RPE脫色素,而黃斑內,特別是中心凹處的RPE保留完好,故熒光造影顯示“牛眼樣"外觀,這種表現常提示患者尚有較好的視力,進展期RP可顯示脈絡膜毛細血管萎縮,此外約25%的病人可出現熒光素滲漏,黃斑區視網膜血管滲漏可伴囊樣黃斑水腫或視網膜變厚而引起中心視力過早損害,熒光素眼底血管造影能幫助RP與無脈絡膜病等其他變性視網膜病的臨床鑒別診斷,而且還有助於對RP少有的並發癥或伴發病,如滲出性視網膜血管病變及黃斑水腫並給予相應的臨床處理,對於防止RP視功能過早喪失十分重要.
5.病理學檢查
臨床上得到的標本均為晚期病例,主要病理改變為視網膜神經上皮層,特別是視桿細胞的進行性退變,繼以視網膜由外向內各層組織的逐漸萎縮,伴發神經膠質增生,色素上皮細胞層亦發生變性和增生,可見色素脫失或積聚,並向視網膜內層遷徙,視網膜血管壁發生玻璃樣變性而增厚,甚至管腔完全閉塞,脈絡膜血管可有不同程度硬化,毛細血管完全或部分消失,視神經可完全萎縮,視盤上常有膠質增生,形成膜狀,與視網膜內的膠質膜相連接,檢眼鏡下所見視盤蠟黃色,一般認為是神經膠質增生所致.
近年來超微結構檢查,已確認視桿狀體外節盤膜在病程早期即已喪失,視錐狀體外節盤膜則相對地有所保留,但也有一些病例在殘存的視錐狀體外節盤膜上,有縮短和空泡等異常改變,推測以上病理改變的原因,可能為結構基因失常,或感光細胞外節盤膜內合成酶及其產物的基因缺陷有關.
6.眼底檢查所見
本病早期雖已有眼底可完全正常,其後,隨病情的進展逐漸出現眼底改變,典型的改變有:
(1)視乳頭萎縮:發生於病程晚期,色淡而略顯黃色,稱為“蠟樣視乳頭",邊緣稍稍模糊,偶有似被一層薄紗遮蓋的朦朧感.
(2)視網膜血管狹窄;血管呈一致性狹窄,尤以動脈為顯著,其狹窄程度反映病的嚴重程度,在晚期,動脈極細,至周邊眼底後即難以辨認而似消失,但無白線化,亦無白鞘包繞.
(3)視網膜色素沉著,始於赤道部,色素呈有突起的小點,繼而增多變大,多呈典型的骨細胞樣,有時呈不規則的線條狀,起初色素斑靠近赤道部呈環形散佈,大多位於視網膜血管附近,特別是靜脈的前面,可遮蓋一部分血管,或沿血管分佈,且多見於血管分支處,以後,色素沉著向中心和周邊部擴展,色素斑的環形散佈區逐漸加寬,甚至佈滿全部眼底,同時視網膜萎縮,色素上皮層色素脫失,暴露出脈絡膜血管而呈豹紋狀眼底,整個眼底灰暗,後期脈絡膜血管亦硬化,呈黃白色條紋,玻璃體一般清晰,有時可見少量點狀或線狀混濁.
(4)FFA:可見脈絡毛細血管萎縮,視網膜血管有閉塞,有時還可見到黃斑,後極部甚至周邊部的熒光滲漏.
原發性視網膜色素變性预防
本病隱性遺傳患者發病早,病情重,發展快,預後極為惡劣,以30歲左右時視功能已高度不良,至50歲左右接近全盲,顯性遺傳患者則反之,偶爾亦有發展至一定程度後趨於靜止者,故預後相對地優於隱性遺傳型,因此可等到勉強正常就學和就業的機會,本病隱性遺傳者,其先輩多有近親聯煙史,禁止近親聯煙可使本病減少發生約22%,另外,隱性遺傳患者應盡量避免與本病傢族史者結婚,更不能與也患有本病者結婚,顯性遺傳患者,其子女發生本病的風險為50%,本病為遺傳性疾病,其先輩多有近親婚姻史,禁止近親婚姻可使本病減少發生約22%.





























