遺傳性共濟失調 G11.904 傢族性共濟失調
遺傳性共濟失調 G11.904 傢族性共濟失調百科
遺傳性共濟失調是一組以慢性進行性小腦性共濟失調為特征的遺傳變性病;有遺傳史,共濟失調表現及小腦損害為主的病理改變是三大特征.本組疾病除小腦及傳導纖維受累外,常累及脊髓後柱,錐體束、腦橋核、基底核,腦神經核、脊神經節和自主神經系統等.共濟失調步態最先出現且逐漸加重,最終使患者臥床,臨床癥狀復雜、交錯重疊,即使同一傢族也可表現高度異質性.
遺傳性共濟失調 G11.904 傢族性共濟失調
遺傳性共濟失調 G11.904 傢族性共濟失調病因
基因遺傳(90%):
小腦性共濟失調(cerebellarataxia,CA)為常染色體顯性遺傳,近年來部分亞型基因已被克隆和測序,顯示致病基因三核苷酸(如CAG)重復序列動態突變,拷貝數逐代增加為致病原因.
常染色體顯性遺傳的脊髓小腦性共濟失調具有遺傳異質性,最具特征性的基因缺陷是擴增的CAG三核苷酸重復編碼多聚谷氨酰胺通道,該通道在功能不明蛋白(ataxins)和神經末梢上發現的P/Q型鈣通道α1A亞單位上;其他類型突變包括CTG三核苷酸(SCA8)和ATTCT五核苷酸(SCA10)重復序列擴增,在許多病例中這種擴增片斷的大小與疾病嚴重性有關,且發病年齡愈小,病情愈重.
Friedreich型共濟失調(FRDA)是9號染色體長臂(9q13-12.1)frataxin基因非編碼區GAA三核苷酸重復序列異常擴增所致,正常GAA重復擴增42次以下,病人異常擴增(66~1700次)形成異常螺旋結構可抑制基因轉錄.
發病機制小腦性共濟失調(ADCA)病理改變主要表現小腦,脊髓和腦幹變性,故又稱為脊髓小腦性共濟失調(SCA),根據臨床特點和基因定位分為SCA1~21種亞型.
SCAs基因突變改變蛋白的性質,使之無法被正常加工,異常加工的片斷與一種參與非溶酶體降解的缺陷蛋白泛素(ubiquitin)結合,共同以蛋白酶體(protease)的復合體形式轉運至核內,推測這種核內蛋白聚集可影響細胞核的功能,每種SCA亞型基因位於不同的染色體,有不同的大小和基因突變部位,例如,SCA1基因位於染色體6q22-23,基因組跨度450Kb,cDNA長11Kb,含9個外顯子,編碼816個氨基酸殘基組成ataxia-1蛋白,該蛋白位於細胞核,CAG突變位於8號外顯子,擴增拷貝數為40~83,正常人為6~38,SCA3(MJD)是我國最常見的SCA亞型,基因位於14q24.3-32,至少含4個外顯子,編碼960個氨基酸殘基組成ataxia-3蛋白,分佈在細胞質中,CAG突變位於4號外顯子,擴增拷貝數為61~89,正常人為12~41.
盡管SCA有共同的基因突變機制,導致各亞型臨床表現雷同,但仍有差異,如有的伴眼肌麻痹,有的伴視網膜色素變性,病理損害部位和程度也不相同,提示除瞭多聚谷氨酰胺毒性作用外,其他因素可能也參與發病.
SCA共同的病理改變主要是小腦,腦幹和脊髓變性和萎縮,但各亞型也有其特點,如SCA1主要是小腦,腦幹的神經元丟失,脊髓小腦束和後索受損,很少累及黑質,基底核及脊髓前角細胞;SCA2以下橄欖核,腦橋,小腦損害為重;SCA3主要損害腦橋和脊髓小腦束;SCA7的特征是視網膜神經細胞變性.
Friedreich型共濟失調(FRDA)基因產物frataxin蛋白存在於脊髓,骨骼肌,心臟及肝臟等細胞線粒體內膜,導致線粒體功能障礙而發病,重復擴增愈多,發病年齡愈早,肉眼可見脊髓變細,胸段明顯;鏡下顯示後索,脊髓小腦束和皮質脊髓束變性,後根神經節和Clarke柱神經元丟失,周圍神經膠質增生,腦幹,小腦和大腦受累較輕,心臟因心肌肥厚而擴大.
遺傳性共濟失調 G11.904 傢族性共濟失調
遺傳性共濟失調 G11.904 傢族性共濟失調症状
依據共濟失調類型的不同分為:
1.脊髓型:⑴Friedreich型共濟失調:
常染色體隱性遺傳,青少年起病,初始行走不穩,漸出現後索損害的癥狀,Romberg征(+),睜眼可以改善.繼之脊髓小腦束受累,出現步基寬,蹣跚步態,定向性震顫和小腦性構音障礙.肢體肌張力降低,腱反射減低或消失,下肢沉重.部分病人可伴有弓形足、脊柱側彎及其他畸形,個別病人可有心臟異常.⑵遺傳性痙攣性截癱:常染色體顯性或隱性遺傳或性連遺傳.兒童期起病,男性多見,主要為錐體束受損,多為下肢呈緩慢加重的痙攣性癱、剪刀狀步態.無感覺障礙,上肢很少受累,可伴有原發性視神經萎縮或視網膜色素變性.
2.小腦型:
⑴Marie型共濟失調:常染色顯性遺傳,成年起病,自下肢開始出現小腦型共濟失調而無感覺障礙,言語常頓挫或暴發,可有錐體束征及欣快,智力減退.
⑵橄欖小腦橋腦萎縮(OPCA):常染色體顯性遺傳,中年後起病,除小腦型共濟失調和構音障礙外有早期尿失禁,部分病人有智能減退和錐體外系癥狀如帕金森綜合征等,但無眼球震顫.
3.脊髓小腦型:
主要有共濟失調-毛細血管擴張癥.常染色體隱性遺傳,嬰兒期發病,小腦型共濟失調,構音障礙,皮膚、顏面毛細血管擴張,多數伴有舞蹈樣手足徐動,隨年齡增長而明顯.青春期後出現深感覺消失等脊髓後索癥狀,和病理反射陽性.可因免疫缺陷而反復發生呼吸道感染,晚期有肺部廣泛纖維化、肺功能不全等.
遺傳性共濟失調 G11.904 傢族性共濟失調
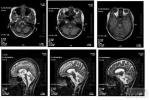
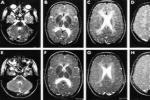
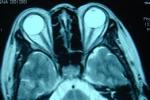
遺傳性共濟失調 G11.904 傢族性共濟失調检查
1.合並弓形足脊柱側彎者相應部位X線攝片有改變.
2.頭顱MRI對OPCA有確診價值.
遺傳性共濟失調 G11.904 傢族性共濟失調预防
應進行遺傳咨詢,預防措施包括避免近親結婚,攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生,本病發展緩慢,如無嚴重的的心肺並發癥,多數不影響壽命,少數患者臥床不起而殘廢.





























